
Rabbit Anti-ADAMTSL2 antibody
ADAMTS like 2; ADAMTS like protein 2; ADAMTS-like protein 2; ADAMTSL 2; ADAMTSL-2; ADAMTSL2; ATL2_HUMAN.
View History [Clear]
Details
Product Name ADAMTSL2 Chinese Name 整合素样金属蛋白酶与凝血酶样2蛋白抗体 Alias ADAMTS like 2; ADAMTS like protein 2; ADAMTS-like protein 2; ADAMTSL 2; ADAMTSL-2; ADAMTSL2; ATL2_HUMAN. Research Area Tumour Cardiovascular Cell biology immunology Signal transduction Growth factors and hormones Cell adhesion molecule Immunogen Species Rabbit Clonality Polyclonal React Species Human, (predicted: Mouse, Rat, ) Applications WB=1:500-2000 ELISA=1:5000-10000
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.Theoretical molecular weight 102kDa Cellular localization Secretory protein Form Liquid Concentration 1mg/ml immunogen KLH conjugated synthetic peptide derived from human ADAMTSL2: 522-580/951 Lsotype IgG Purification affinity purified by Protein A Buffer Solution 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Storage Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. Attention This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. PubMed PubMed Product Detail ADAMTS (A Disintegrin And Metalloproteinase Domain with Thrombospondin type 1 Modules) is a family of zinc-dependent proteases that are implicated in a variety of normal and pathological conditions, including arthritis and cancer. ADAMTS protein family members contain an amino-terminal propeptide domain, a metalloproteinase domain, a disintegrin-like domain and a carboxy-terminus that contains a varying number of Thrombospondin type 1 (TSP-1) motifs. ADAMTS-L2 (ADAMTS-like protein 2) is a 951 amino acid secreted protein that is highly expressed in lung, kidney and liver. Mutations in the gene encoding ADAMTS are the cause of geleophysic dysplasia, an autosomal recessive disorder characterized by cardiac vavular anomalies, short stature, thick skin and brachydactyly. In individuals affected with geleophysic dysplasia, there is a significant increase in total active TGF-beta 1 and nuclear locations of p-SAMD2 in fibroblasts. Interestingly, ADAMTS-L2 interacts with LTBP-1, a glycoprotein that is part of the platelet-derived TGF-beta 1 complex.
Function:
Defects in ADAMTSL2 are the cause of geleophysic dysplasia [MIM:231050]. Geleophysic dysplasia is an autosomal recessive disorder characterized by short stature, brachydactyly, thick skin and cardiac valvular anomalies often responsible for an early death.
Subcellular Location:
Secreted.
Post-translational modifications:
Glycosylated (By similarity). Can be O-fucosylated by POFUT2 on a serine or a threonine residue found within the consensus sequence C1-X(2)-(S/T)-C2-G of the TSP type-1 repeat domains where C1 and C2 are the first and second cysteine residue of the repeat, respectively. Fucosylated repeats can then be further glycosylated by the addition of a beta-1,3-glucose residue by the glucosyltransferase, B3GALTL. Fucosylation mediates the efficient secretion of ADAMTS family members. Also can be C-glycosylated with one or two mannose molecules on tryptophan residues within the consensus sequence W-X-X-W of the TPRs, and N-glycosylated. These other glycosylations can also facilitate secretion (By similarity).
DISEASE:
Defects in ADAMTSL2 are the cause of geleophysic dysplasia (GLPD) [MIM:231050]. Geleophysic dysplasia is an autosomal recessive disorder characterized by short stature, brachydactyly, thick skin and cardiac valvular anomalies often responsible for an early death.
Similarity:
Contains 1 PLAC domain.
Contains 7 TSP type-1 domains.
SWISS:
Q86TH1
Gene ID:
9719
Database links:Entrez Gene: 9719 Human
Entrez Gene: 77794 Mouse
Omim: 612277 Human
SwissProt: Q86TH1 Human
SwissProt: Q7TSK7 Mouse
Unigene: 522543 Human
Unigene: 330088 Mouse
Extracellular matrix蛋白Product Picture 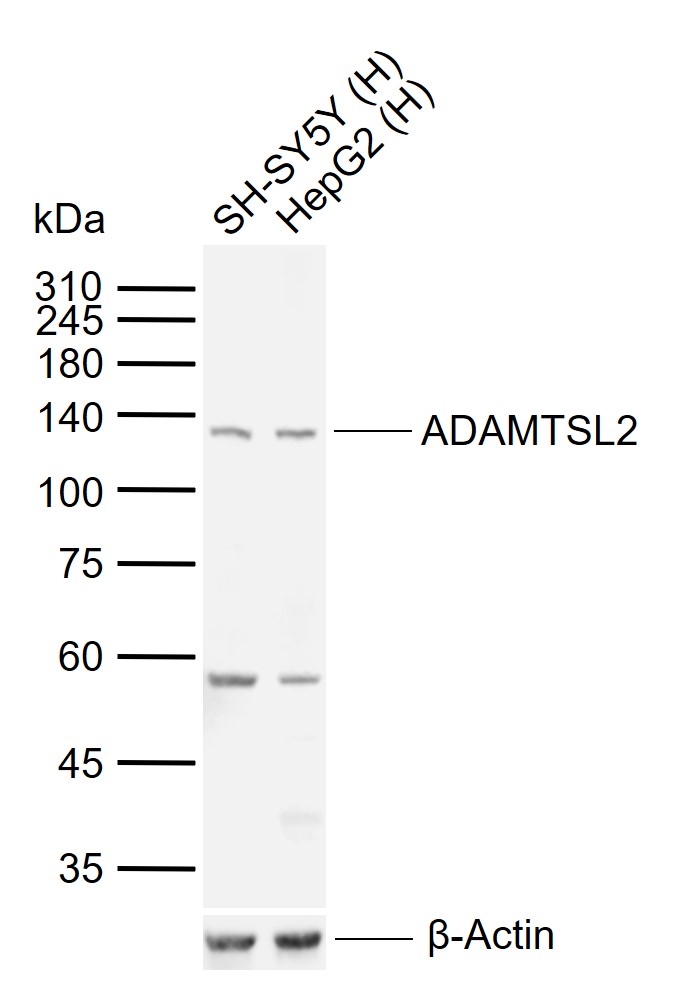 Sample:
Sample:
Lane 1: Human SH-SY5Y cell lysates
Lane 2: Human HepG2 cell lysates
Primary: Anti-ADAMTSL2 (SL5862R) at 1/1000 dilution
Secondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilution
Predicted band size: 102 kDa
Observed band size: 135 kDa
Cartpieces
Totalgoods,subtotals:¥Checkout
References (0)
No References
Bought notes(bought amounts latest0)
No one bought this product
User Comment(Total0User Comment Num)
- No comment


 +86 571 56623320
+86 571 56623320
 +86 18668110335
+86 18668110335

