
Rabbit Anti-Beta galactosidase antibody
BGAL_HUMAN; Beta-galactosidase; GLB1; EC:3.2.1.23; Acid beta-galactosidase (Lactase); Elastin receptor 1; ELNR1; galactosidase beta 1; EBP; ELNR1; MPS4B; β-Galactosidase; β Galactosidase.
View History [Clear]
Details
Product Name Beta galactosidase Chinese Name β半乳糖苷酶抗体 Alias BGAL_HUMAN; Beta-galactosidase; GLB1; EC:3.2.1.23; Acid beta-galactosidase (Lactase); Elastin receptor 1; ELNR1; galactosidase beta 1; EBP; ELNR1; MPS4B; β-Galactosidase; β Galactosidase. literatures Research Area immunology Immunogen Species Rabbit Clonality Polyclonal React Species Human, Mouse, Rat, (predicted: Dog, ) Applications IHC-P=1:100-500 IF=1:100-500 (Paraffin sections need antigen repair)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.Theoretical molecular weight 71kDa Cellular localization cytoplasmic Form Liquid Concentration 1mg/ml immunogen KLH conjugated synthetic peptide derived from human Beta galactosidase: 221-290/677 Lsotype IgG Purification affinity purified by Protein A Buffer Solution 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Storage Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. Attention This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. PubMed PubMed Product Detail This gene encodes a member of the glycosyl hydrolase 35 family of proteins. Alternative splicing results in multiple transcript variants, at least one of which encodes a preproprotein that is proteolytically processed to generate the mature lysosomal enzyme. This enzyme catalyzes the hydrolysis of a terminal beta-linked galactose residue from ganglioside substrates and other glycoconjugates. Mutations in this gene may result in GM1-gangliosidosis and Morquio B syndrome. [provided by RefSeq, Nov 2015]
Function:
Cleaves beta-linked terminal galactosyl residues from gangliosides, glycoproteins, and glycosaminoglycans.
Isoform 2 has no beta-galactosidase catalytic activity, but plays functional roles in the formation of extracellular elastic fibers (elastogenesis) and in the development of connective tissue. Seems to be identical to the elastin-binding protein (EBP), a major component of the non-integrin cell surface receptor expressed on fibroblasts, smooth muscle cells, chondroblasts, leukocytes, and certain cancer cell types. In elastin producing cells, associates with tropoelastin intracellularly and functions as a recycling molecular chaperone which facilitates the secretions of tropoelastin and its assembly into elastic fibers.
Subcellular Location:
Isoform 1: Lysosome. Isoform 2: Cytoplasm, perinuclear region. Note=Localized to the perinuclear area of the cytoplasm but not to lysosomes.
DISEASE:
Defects in GLB1 are the cause of GM1-gangliosidosis type 1 (GM1G1) [MIM:230500]; also known as infantile GM1-gangliosidosis. GM1-gangliosidosis is an autosomal recessive lysosomal storage disease marked by the accumulation of GM1 gangliosides, glycoproteins and keratan sulfate primarily in neurons of the central nervous system. GM1G1 is characterized by onset within the irst three months of life, central nervous system degeneration, coarse facial features, hepatosplenomegaly, skeletal dysmorphology reminiscent of Hurler syndrome, and rapidly progressive psychomotor deterioration. Urinary oligosaccharide levels are high. It leads to death usually between the first and second year of life.
Defects in GLB1 are the cause of GM1-gangliosidosis type 2 (GM1G2) [MIM:230600]; also known as late infantile/juvenile GM1-gangliosidosis. GM1G2 is characterized by onset between ages 1 and 5. The main symptom is locomotor ataxia, ultimately leading to a state of decerebration with epileptic seizures. Patients do not display the skeletal changes associated with the infantile form, but they nonetheless excrete elevated amounts of beta-linked galactose-terminal oligosaccharides. Inheritance is autosomal recessive.
Defects in GLB1 are the cause of GM1-gangliosidosis type 3 (GM1G3) [MIM:230650]; also known as adult or chronic GM1-gangliosidosis. GM1G3 is characterized by a variable phenotype. Patients show mild skeletal abnormalities, dysarthria, gait disturbance, dystonia and visual impairment. Visceromegaly is absent. Intellectual deficit can initially be mild or absent but progresses over time. Inheritance is autosomal recessive.
Defects in GLB1 are the cause of mucopolysaccharidosis type 4B (MPS4B) [MIM:253010]; also known as Morquio syndrome B. MPS4B is a form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life.
Similarity:
Belongs to the glycosyl hydrolase 35 family.
SWISS:
P16278
Gene ID:
2720
Database links:
Product Picture 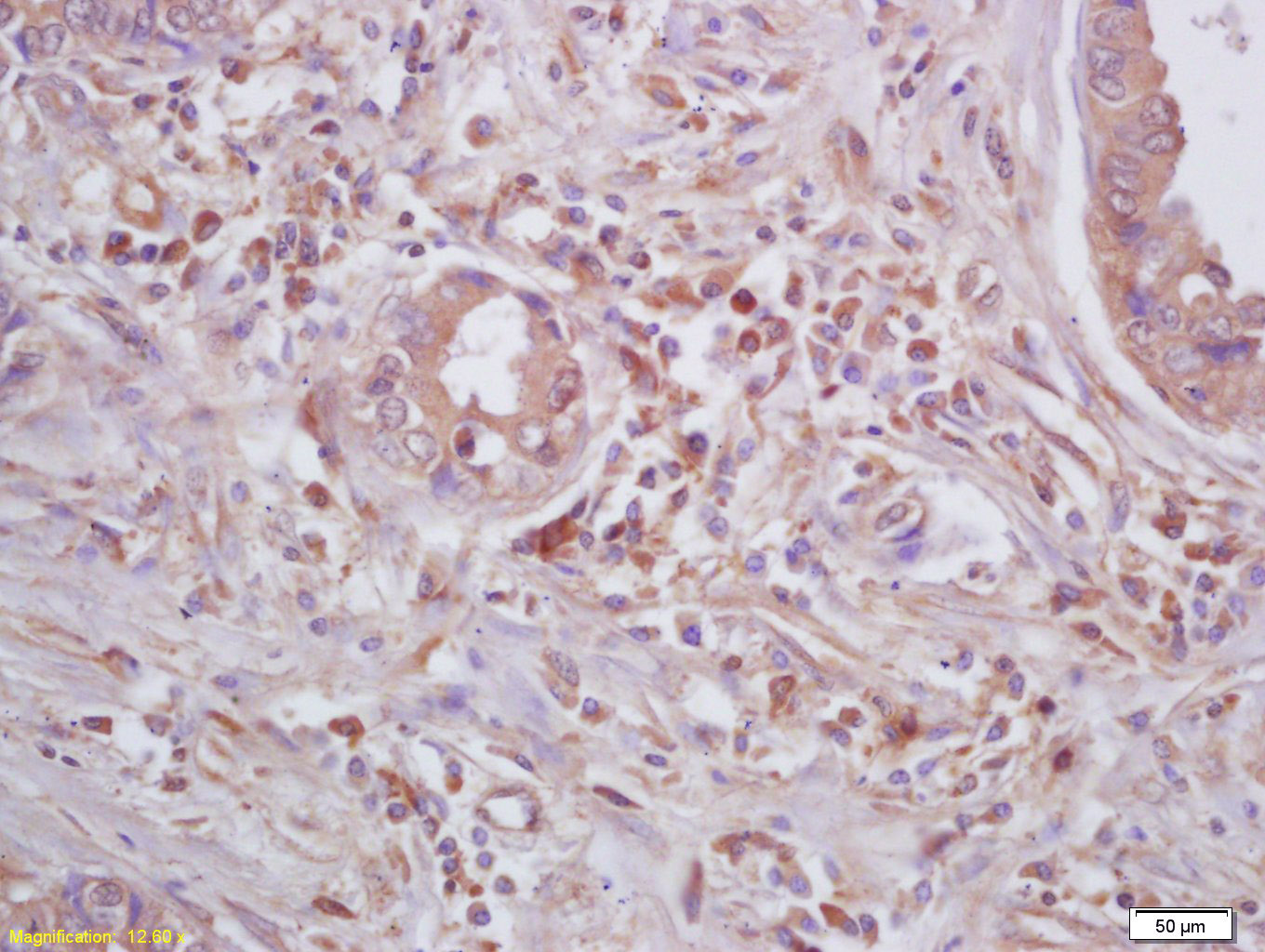 Tissue/cell: human lung carcinoma; 4% Paraformaldehyde-fixed and paraffin-embedded;
Tissue/cell: human lung carcinoma; 4% Paraformaldehyde-fixed and paraffin-embedded;
Antigen retrieval: citrate buffer ( 0.01M, pH 6.0 ), Boiling bathing for 15min; Block endogenous peroxidase by 3% Hydrogen peroxide for 30min; Blocking buffer (normal goat serum,C-0005) at 37℃ for 20 min;
Incubation: Anti-Beta galactosidase Polyclonal Antibody, Unconjugated(SL4631R) 1:200, overnight at 4°C, followed by conjugation to the secondary antibody(SP-0023) and DAB(C-0010) staining
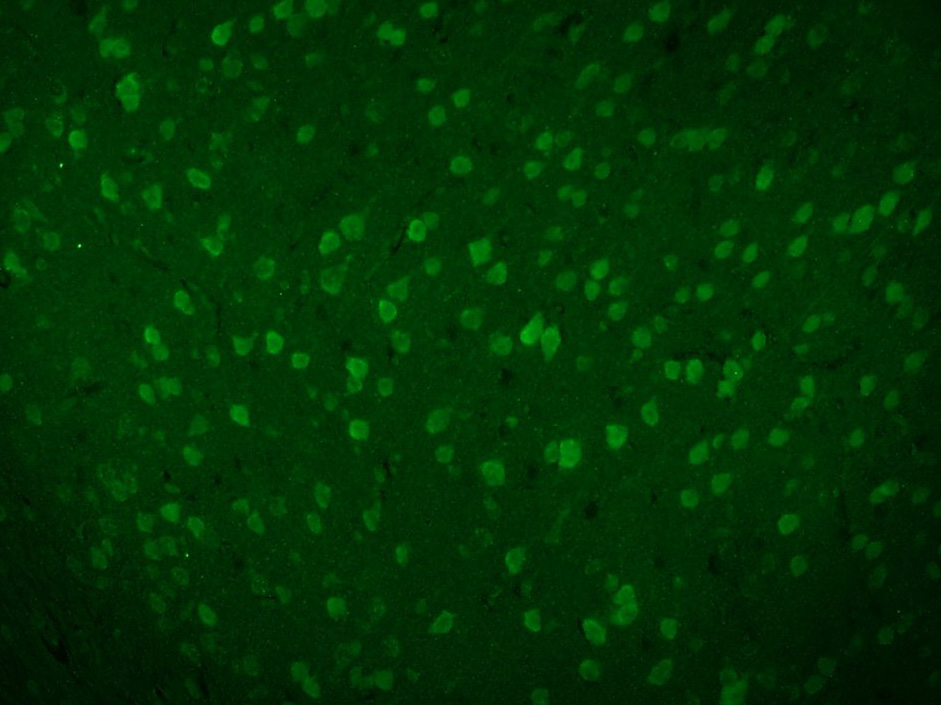 Paraformaldehyde-fixed, paraffin embedded (Mouse brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (Beta galactosidase) Polyclonal Antibody, Unconjugated (SL4631R) at 1:200 overnight at 4°C, followed by a conjugated Goat Anti-Rabbit IgG antibody (SL0295G-AF488) for 90 minutes, and DAPI for nuclei staining.
Paraformaldehyde-fixed, paraffin embedded (Mouse brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (Beta galactosidase) Polyclonal Antibody, Unconjugated (SL4631R) at 1:200 overnight at 4°C, followed by a conjugated Goat Anti-Rabbit IgG antibody (SL0295G-AF488) for 90 minutes, and DAPI for nuclei staining.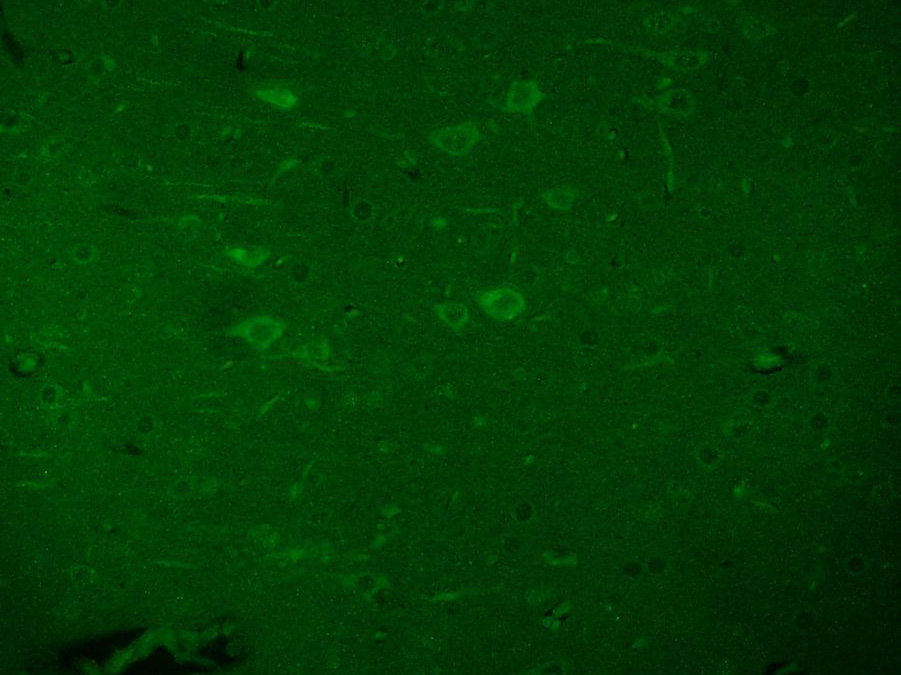 Paraformaldehyde-fixed, paraffin embedded (Rat brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (Beta galactosidase) Polyclonal Antibody, Unconjugated (SL4631R) at 1:200 overnight at 4°C, followed by a conjugated Goat Anti-Rabbit IgG antibody (SL0295G-AF488) for 90 minutes, and DAPI for nuclei staining.
Paraformaldehyde-fixed, paraffin embedded (Rat brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (Beta galactosidase) Polyclonal Antibody, Unconjugated (SL4631R) at 1:200 overnight at 4°C, followed by a conjugated Goat Anti-Rabbit IgG antibody (SL0295G-AF488) for 90 minutes, and DAPI for nuclei staining.
Cartpieces
Totalgoods,subtotals:¥Checkout
References (0)
No References
Bought notes(bought amounts latest0)
No one bought this product
User Comment(Total0User Comment Num)
- No comment


 +86 571 56623320
+86 571 56623320
 +86 18668110335
+86 18668110335

