
Rabbit Anti-LRP5 antibody
BMND1; HBM; HGNC:6697; Low density lipoprotein receptor related protein 5; Low density lipoprotein receptor related protein 7; LR3; LRP7; OPPG; OPS; LRP7; Osteoporosis pseudoglioma syndrome; VBCH2; LRP5_HUMAN .
View History [Clear]
Details
Product Name LRP5 Chinese Name 低密度Lipoprotein受体相关蛋白5抗体 Alias BMND1; HBM; HGNC:6697; Low density lipoprotein receptor related protein 5; Low density lipoprotein receptor related protein 7; LR3; LRP7; OPPG; OPS; LRP7; Osteoporosis pseudoglioma syndrome; VBCH2; LRP5_HUMAN . Research Area Cardiovascular Cell biology immunology Neurobiology Signal transduction Stem cells Immunogen Species Rabbit Clonality Polyclonal React Species Human, Mouse, (predicted: Rat, Chicken, Dog, Cow, Horse, Rabbit, ) Applications ELISA=1:5000-10000 IHC-P=1:100-500 IHC-F=1:100-500 ICC=1:100 IF=1:100-500 (Paraffin sections need antigen repair)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.Theoretical molecular weight 176kDa Cellular localization cytoplasmic The cell membrane Form Liquid Concentration 1mg/ml immunogen KLH conjugated synthetic peptide derived from human LRP5: 501-600/1615 <Extracellular> Lsotype IgG Purification affinity purified by Protein A Buffer Solution 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Storage Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. Attention This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. PubMed PubMed Product Detail LRP5 is involved in the Wnt/beta catenin signaling pathway, probably by acting as a coreceptor together with Frizzled for Wnt. Defects in LRP5 are a cause of autosomal dominant and autosomal recessive familial exudative vitreoretinopathy (FEVR). Autosomal dominant FEVR is also referred to as exudative vitreoretinopathy 1 (EVR1); also known as Criswick-Schepens syndrome. FEVR is a disorder of the retinal vasculature characterized by an abrupt cessation of growth of peripheral capillaries, leading to an avascular peripheral retina. This may lead to compensatory retinal neovascularization, which is thought to be induced by hypoxia from the initial avascular insult. New vessels are prone to leakage and rupture causing exudates and bleeding, followed by scarring, retinal detachment and blindness. FEVR is reported to have a penetrance of 100%, but clinical features can be highly variable, even within the same family. Patients with mild forms of the disease are asymptomatic, and their only disease-related abnormality is an arc of avascular retina in the extreme temporal periphery.
Function:
Component of the Wnt-Fzd-LRP5-LRP6 complex that triggers beta-catenin signaling through inducing aggregation of receptor-ligand complexes into ribosome-sized signalsomes. Cell-surface coreceptor of Wnt/beta-catenin signaling, which plays a pivotal role in bone formation. The Wnt-induced Fzd/LRP6 coreceptor complex recruits DVL1 polymers to the plasma membrane which, in turn, recruits the AXIN1/GSK3B-complex to the cell surface promoting the formation of signalsomes and inhibiting AXIN1/GSK3-mediated phosphorylation and destruction of beta-catenin. Appears be required for postnatal control of vascular regression in the eye. Required for posterior patterning of the epiblast during gastrulation.
Subunit:
Homodimer; disulfide-linked. Forms phosphorylated oligomer aggregates on Wnt-signaling (By similarity). Component of a Wnt-signaling complex that contains a WNT protein, a FZD protein and LRP5 or LRP6. Interacts with FZD8; the interaction is formed on WNT-binding and signaling. Interacts (via the phosphorylated PPPSP motif domains) with AXIN1; the interaction prevents inhibition of beta-catenin phosphorylation and signaling and is enhanced in the presence of GSK3B and WNT1 or WNT3A. Interacts (via beta-propeller regions 3 and 4) with DKK1; the interaction, enhanced by MESD and/or KREMEN, inhibits beta-catenin signaling by preventing GSK3-mediated phosphorylation of the PPPSP motifs and subsequent, AXIN1 binding. Interacts with MESD; the interaction prevents the formation of LRP5 aggregates, targets LRP5 to the plasma membrane and, when complexed with KREMEN2, increases DKK1 binding. Interacts with CSNK1E. Interacts with SOST; the interaction antagonizes canonical Wnt signaling. Interacts with APCDD1.
Subcellular Location:
Membrane; Single-pass type I membrane protein. Endoplasmic reticulum.
Tissue Specificity:
Widely expressed, with the highest level of expression in the liver.
Post-translational modifications:
Phosphorylation of cytoplasmic PPPSP motifs regulates the signal transduction of the Wnt signaling pathway through acting as a docking site for AXIN1 (By similarity).
DISEASE:
Defects in LRP5 are the cause of vitreoretinopathy exudative type 4 (EVR4) [MIM:601813]. EVR4 is a disorder of the retinal vasculature characterized by an abrupt cessation of growth of peripheral capillaries, leading to an avascular peripheral retina. This may lead to compensatory retinal neovascularization, which is thought to be induced by hypoxia from the initial avascular insult. New vessels are prone to leakage and rupture causing exudates and bleeding, followed by scarring, retinal detachment and blindness. Clinical features can be highly variable, even within the same family. Patients with mild forms of the disease are asymptomatic, and their only disease related abnormality is an arc of avascular retina in the extreme temporal periphery. EVR4 inheritance can be autosomal dominant or recessive.
Genetic variations in LRP5 are a cause of susceptibility to osteoporosis (OSTEOP) [MIM:166710]; also known as senile osteoporosis or postmenopausal osteoporosis. Osteoporosis is characterized by reduced bone mass, disruption of bone microarchitecture without alteration in the composition of bone. Osteoporotic bones are more at risk of fracture.
Defects in LRP5 are the cause of osteoporosis-pseudoglioma syndrome (OPPG) [MIM:259770]; also known as osteogenesis imperfecta ocular form. OPPG is a recessive disorder characterized by very low bone mass and blindness. Individualy with OPPG are prone to develop bone fractures and deformations and have various eye abnormalities, including phthisis bulbi, retinal detachments, falciform folds or persistent vitreal vasculature.
Defects in LRP5 are a cause of high bone mass trait (HBM) [MIM:601884]. HBM is a rare phenotype characterized by exceptionally dense bones. HBM individuals show otherwise a completely normal skeletal structure and no other unusual clinical findings.
Defects in LRP5 are a cause of endosteal hyperostosis Worth type (WENHY) [MIM:144750]; also known as autosomal dominant osteosclerosis. WENHY is an autosomal dominant sclerosing bone dysplasia clinically characterized by elongation of the mandible, increased gonial angle, flattened forehead, and the presence of a slowly enlarging osseous prominence of the hard palate (torus palatinus). Serum calcium, phosphorus and alkaline phosphatase levels are normal. Radiologically, it is characterized by early thickening of the endosteum of long bones, the skull and of the mandible. With advancing age, the trabeculae of the metaphysis become thickened. WENHY becomes clinically and radiologically evident by adolescence, does not cause deformity except in the skull and mandible, and is not associated with bone pain or fracture. Affected patients have normal height, proportion, intelligence and longevity.
Defects in LRP5 are the cause of osteopetrosis autosomal dominant type 1 (OPTA1) [MIM:607634]. Osteopetrosis is a rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. The disorder occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. OPTA1 is characterized by generalized osteosclerosis most pronounced in the cranial vault. Patients are often asymptomatic, but some suffer from pain and hearing loss. It appears to be the only type of osteopetrosis not associated with an increased fracture rate.
Defects in LRP5 are the cause of van Buchem disease type 2 (VBCH2)[MIM:607636]. VBCH2 is an autosomal dominant sclerosing bone dysplasia characterized by cranial osteosclerosis, thickened calvaria and cortices of long bones, enlarged mandible and normal serum alkaline phosphatase levels.
Similarity:
Belongs to the LDLR family.
Contains 4 EGF-like domains.
Contains 3 LDL-receptor class A domains.
Contains 20 LDL-receptor class B repeats.
SWISS:
O75197
Gene ID:
4041
Database links:Entrez Gene: 4041 Human
Entrez Gene: 16973 Mouse
Omim: 603506 Human
SwissProt: O75197 Human
SwissProt: Q91VN0 Mouse
Unigene: 6347 Human
Unigene: 274581 Mouse
Product Picture 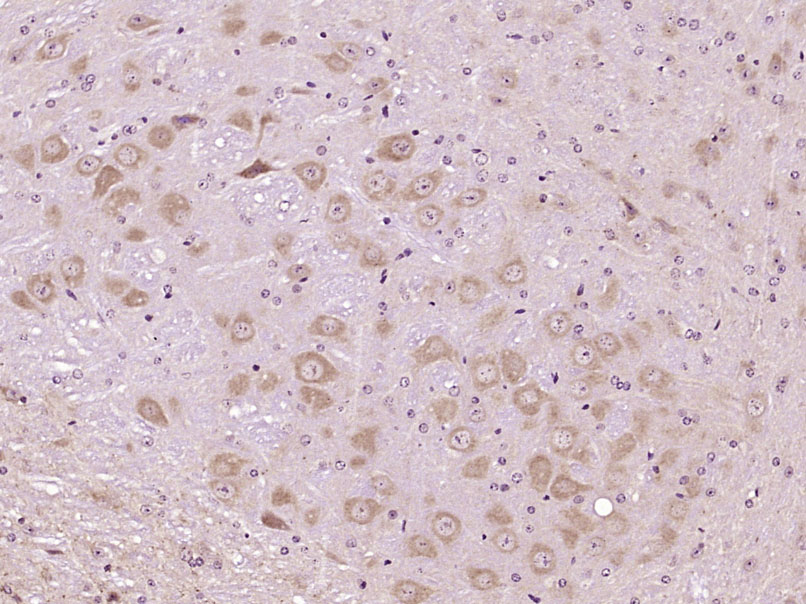 Paraformaldehyde-fixed, paraffin embedded (Mouse brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (LRP5) Polyclonal Antibody, Unconjugated (SL4117R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.
Paraformaldehyde-fixed, paraffin embedded (Mouse brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (LRP5) Polyclonal Antibody, Unconjugated (SL4117R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.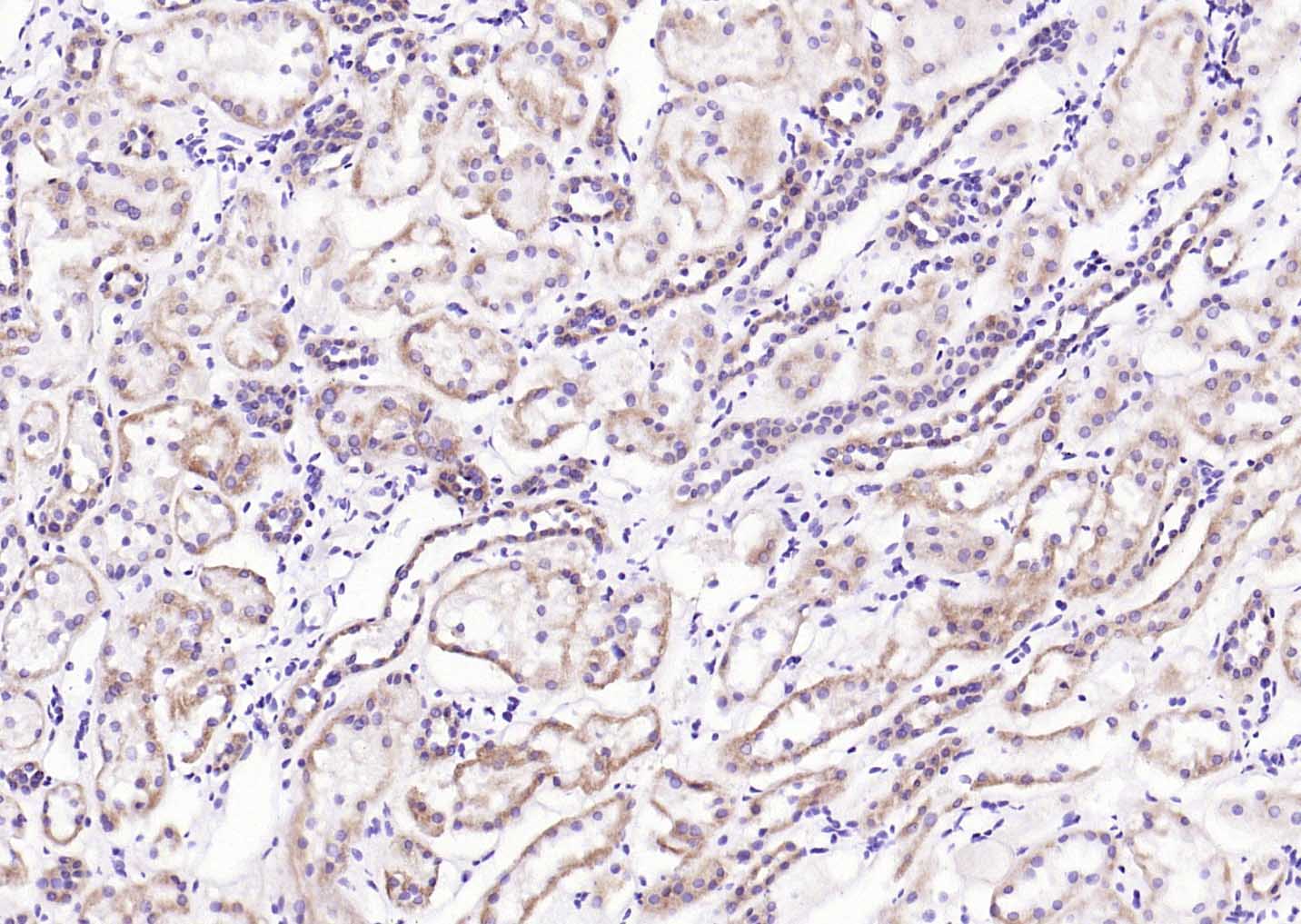 Paraformaldehyde-fixed, paraffin embedded (Human kidney); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (LRP5) Polyclonal Antibody, Unconjugated (SL4117R) at 1:200 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.
Paraformaldehyde-fixed, paraffin embedded (Human kidney); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (LRP5) Polyclonal Antibody, Unconjugated (SL4117R) at 1:200 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.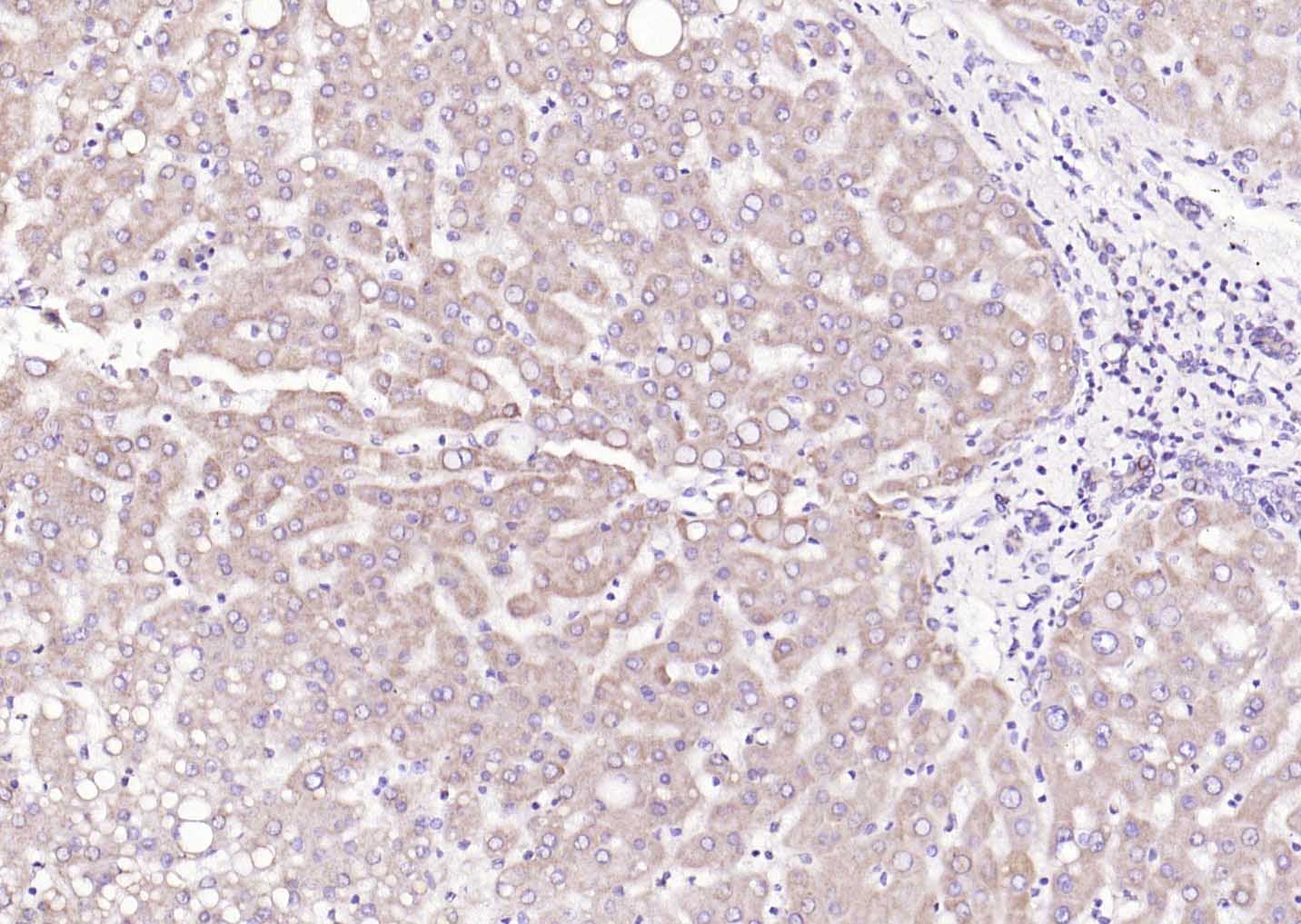 Paraformaldehyde-fixed, paraffin embedded (human liver); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (LRP5) Polyclonal Antibody, Unconjugated (SL4117R) at 1:200 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.
Paraformaldehyde-fixed, paraffin embedded (human liver); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (LRP5) Polyclonal Antibody, Unconjugated (SL4117R) at 1:200 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.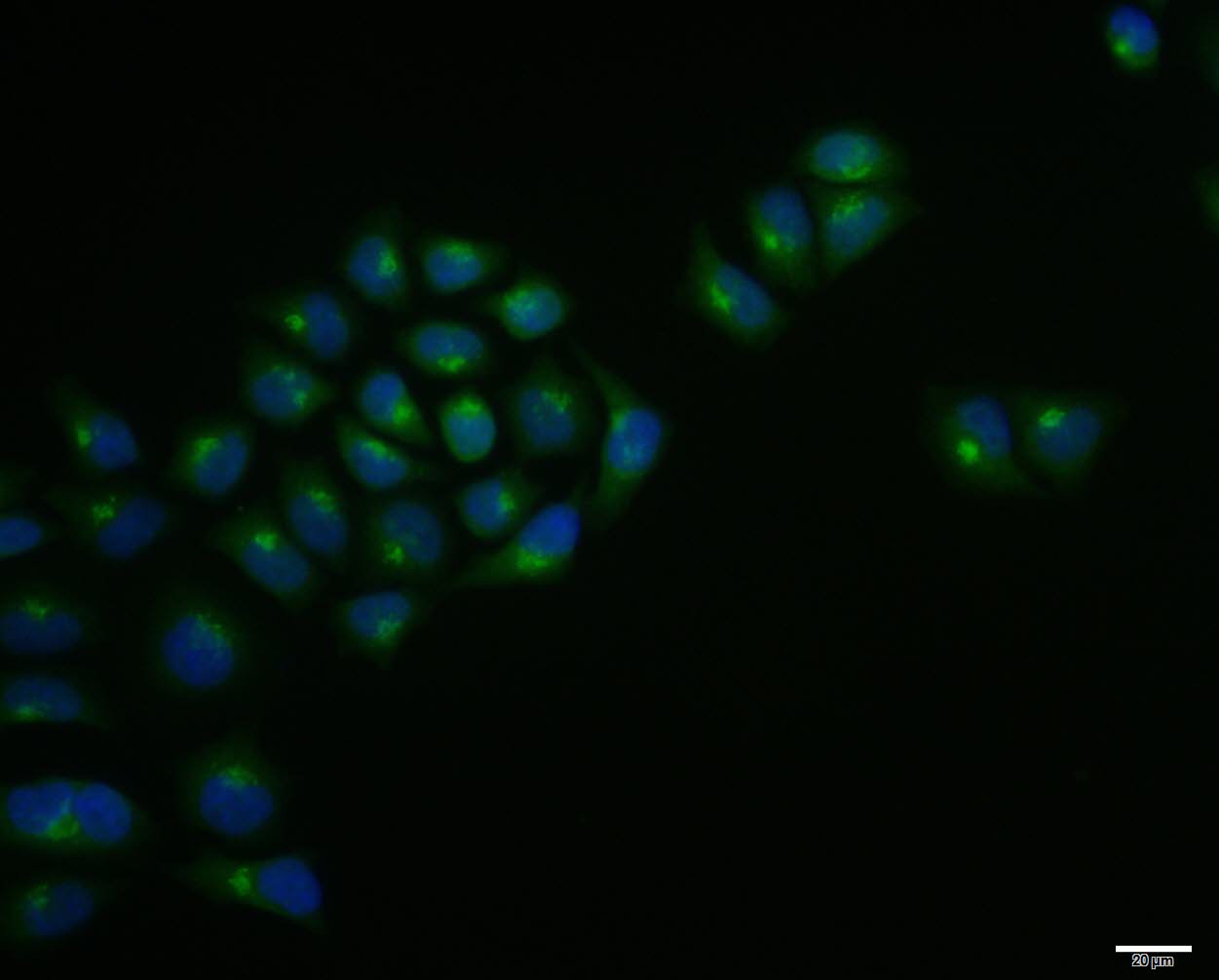 Hela cell; 4% Paraformaldehyde-fixed; Triton X-100 at room temperature for 20 min; Blocking buffer (normal goat serum, C-0005) at 37°C for 20 min; Antibody incubation with (LRP5) polyclonal Antibody, Unconjugated (SL4117R) 1:100, 90 minutes at 37°C; followed by a conjugated Goat Anti-Rabbit IgG antibody at 37°C for 90 minutes, DAPI (blue, C02-04002) was used to stain the cell nuclei.
Hela cell; 4% Paraformaldehyde-fixed; Triton X-100 at room temperature for 20 min; Blocking buffer (normal goat serum, C-0005) at 37°C for 20 min; Antibody incubation with (LRP5) polyclonal Antibody, Unconjugated (SL4117R) 1:100, 90 minutes at 37°C; followed by a conjugated Goat Anti-Rabbit IgG antibody at 37°C for 90 minutes, DAPI (blue, C02-04002) was used to stain the cell nuclei.
Cartpieces
Totalgoods,subtotals:¥Checkout
References (0)
No References
Bought notes(bought amounts latest0)
No one bought this product
User Comment(Total0User Comment Num)
- No comment


 +86 571 56623320
+86 571 56623320
 +86 18668110335
+86 18668110335

