
Rabbit Anti-PAH antibody
PH; PH4H_HUMAN; Phe 4 monooxygenase; Phe-4-monooxygenase; Phenylalanine 4 hydroxylase; Phenylalanine hydroxylase; Phenylalanine-4-hydroxylase; PKU; PKU1.
View History [Clear]
Details
Product Name PAH Chinese Name 苯丙氨酸羟化酶4抗体 Alias PH; PH4H_HUMAN; Phe 4 monooxygenase; Phe-4-monooxygenase; Phenylalanine 4 hydroxylase; Phenylalanine hydroxylase; Phenylalanine-4-hydroxylase; PKU; PKU1. Research Area Tumour Cell biology Signal transduction Stem cells Kinases and Phosphatases Immunogen Species Rabbit Clonality Polyclonal React Species Human, (predicted: Mouse, Rat, Cow, ) Applications WB=1:500-2000
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.Theoretical molecular weight 52kDa Cellular localization cytoplasmic Form Liquid Concentration 1mg/ml immunogen KLH conjugated synthetic peptide derived from human PAH: 1-100/452 Lsotype IgG Purification affinity purified by Protein A Buffer Solution 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Storage Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. Attention This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. PubMed PubMed Product Detail This gene encodes a member of the biopterin-dependent aromatic amino acid hydroxylase protein family. The encoded phenylalanine hydroxylase enzyme hydroxylates phenylalanine to tyrosine and is the rate-limiting step in phenylalanine catabolism. Deficiency of this enzyme activity results in the autosomal recessive disorder phenylketonuria. [provided by RefSeq, Aug 2017]
Subunit:
Homodimer and homotetramer.
Tissue Specificity:
Optimum temperature is 50 degrees Celsius.
DISEASE:
Defects in PAH are the cause of phenylketonuria (PKU) [MIM:261600]. PKU is an autosomal recessive inborn error of phenylalanine metabolism, due to severe phenylalanine hydroxylase deficiency. It is characterized by blood concentrations of phenylalanine persistently above 1200 mumol (normal concentration 100 mumol) which usually causes mental retardation (unless low phenylalanine diet is introduced early in life). They tend to have light pigmentation, rashes similar to eczema, epilepsy, extreme hyperactivity, psychotic states and an unpleasant 'mousy' odor.
Defects in PAH are the cause of non-phenylketonuria hyperphenylalaninemia (Non-PKU HPA) [MIM:261600]. Non-PKU HPA is a mild form of phenylalanine hydroxylase deficiency characterized by phenylalanine levels persistently below 600 mumol, which allows normal intellectual and behavioral development without treatment. Non-PKU HPA is usually caused by the combined effect of a mild hyperphenylalaninemia mutation and a severe one.
Defects in PAH are the cause of hyperphenylalaninemia (HPA) [MIM:261600]. HPA is the mildest form of phenylalanine hydroxylase deficiency.
Similarity:
Belongs to the biopterin-dependent aromatic amino acid hydroxylase family. Contains 1 ACT domain.
SWISS:
P00439
Gene ID:
5053
Database links:Entrez Gene: 408024 Chicken
Entrez Gene: 5053 Human
Entrez Gene: 18478 Mouse
Omim: 612349 Human
SwissProt: P00439 Human
SwissProt: P16331 Mouse
Unigene: 603740 Human
Unigene: 263539 Mouse
Unigene: 1652 Rat
Product Picture 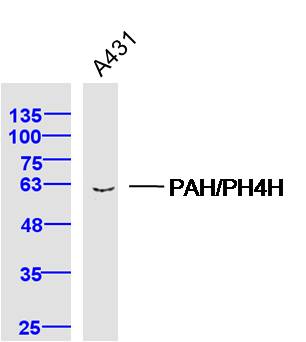 Sample: A431 Cell (Human) Lysate at 40 ug
Sample: A431 Cell (Human) Lysate at 40 ug
Primary: Anti-PAH/PH4H (SL21005R) at 1/300 dilution
Secondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilution
Predicted band size: 52 kD
Observed band size: 60 kD
Cartpieces
Totalgoods,subtotals:¥Checkout
References (0)
No References
Bought notes(bought amounts latest0)
No one bought this product
User Comment(Total0User Comment Num)
- No comment


 +86 571 56623320
+86 571 56623320
 +86 18668110335
+86 18668110335

