
Rabbit Anti-IDUA antibody
IDUA_HUMAN; Alpha-L-iduronidase; IDA; Iduronidase alpha L; MPS1.
View History [Clear]
Details
Product Name IDUA Chinese Name α-L-艾杜糖苷酶抗体 Alias IDUA_HUMAN; Alpha-L-iduronidase; IDA; Iduronidase alpha L; MPS1. Research Area Tumour Cell biology immunology Signal transduction Cell type markers Cytoskeleton Immunogen Species Rabbit Clonality Polyclonal React Species Human, Mouse, Applications WB=1:500-2000 ELISA=1:5000-10000
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.Theoretical molecular weight 70kDa Cellular localization cytoplasmic Form Liquid Concentration 1mg/ml immunogen KLH conjugated synthetic peptide derived from human IDUA: 101-200/653 Lsotype IgG Purification affinity purified by Protein A Buffer Solution 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Storage Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. Attention This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. PubMed PubMed Product Detail This gene encodes an enzyme that hydrolyzes the terminal alpha-L-iduronic acid residues of two glycosaminoglycans, dermatan sulfate and heparan sulfate. This hydrolysis is required for the lysosomal degradation of these glycosaminoglycans. Mutations in this gene that result in enzymatic deficiency lead to the autosomal recessive disease mucopolysaccharidosis type I (MPS I). [provided by RefSeq, Jul 2008].
Subunit:
Monomer (Probable).
Subcellular Location:
Lysosome.
Tissue Specificity:
Ubiquitous.
DISEASE:
Mucopolysaccharidosis 1H (MPS1H) [MIM:607014]: A severe form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1H usually present, within the first year of life, a combination of hepatosplenomegaly, skeletal deformities, corneal clouding and severe mental retardation. Obstructive airways disease, respiratory infection and cardiac complications usually result in death before 10 years of age. Note=The disease is caused by mutations affecting the gene represented in this entry.
Mucopolysaccharidosis 1H/S (MPS1H/S) [MIM:607015]: A form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. MPS1H/S represents an intermediate phenotype of the MPS1 clinical spectrum. It is characterized by relatively little neurological involvement, but most of the somatic symptoms described for severe MPS1 develop in the early to mid-teens, causing considerable loss of mobility. Note=The disease is caused by mutations affecting the gene represented in this entry.
Mucopolysaccharidosis 1S (MPS1S) [MIM:607016]: A mild form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1S may have little or no neurological involvement, normal stature and life span, but present development of joints stiffness, mild hepatosplenomegaly, aortic valve disease and corneal clouding. Note=The disease is caused by mutations affecting the gene represented in this entry.
Similarity:
Belongs to the glycosyl hydrolase 39 family.
SWISS:
P35475
Gene ID:
3425
Database links:Entrez Gene: 3425 Human
Entrez Gene: 15932 Mouse
Omim: 252800 Human
SwissProt: P35475 Human
SwissProt: P48441 Mouse
Unigene: 89560 Human
Product Picture 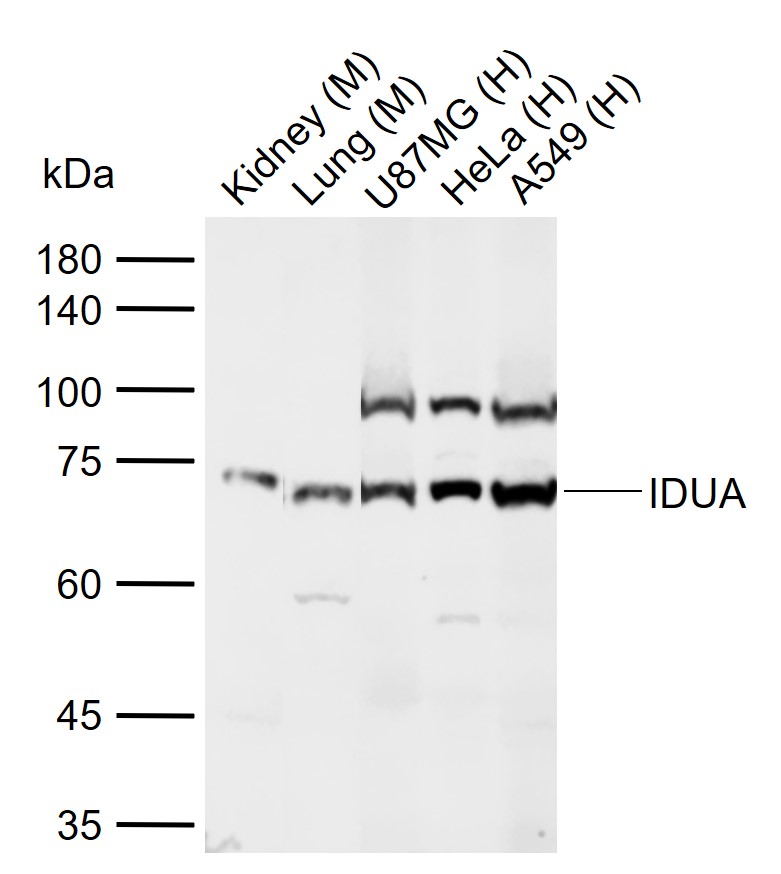 Sample:
Sample:
Lane 1: Mouse Kidney tissue lysates
Lane 2: Mouse Lung tissue lysates
Lane 3: Human U87MG cell lysates
Lane 4: Human HeLa cell lysates
Lane 5: Human A549 cell lysates
Primary: Anti-IDUA (SL15542R) at 1/1000 dilution
Secondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilution
Predicted band size: 70 kDa
Observed band size: 70 kDa
Cartpieces
Totalgoods,subtotals:¥Checkout
References (0)
No References
Bought notes(bought amounts latest0)
No one bought this product
User Comment(Total0User Comment Num)
- No comment


 +86 571 56623320
+86 571 56623320
 +86 18668110335
+86 18668110335

