
Mouse Anti-CK14 (ready to use)antibody (BH0073)
Cytokeratin 14; CK 14; Cytokeratin14; Cytokeratin-14; Dowling Meara; ebs3; Epidermolysis bullosa simplex; k14; Keratin 14; Keratin type I cytoskeletal 14; Keratin14; Koebner; Krt 14; krt14; EBS3; EBS4; NFJ; Keratin, type I cytoskeletal 14; K1C14_HUMAN.
View History [Clear]
Details
Product Name CK14(ready to use) Chinese Name 细胞角蛋白14单克隆抗体(工作液) Alias Cytokeratin 14; CK 14; Cytokeratin14; Cytokeratin-14; Dowling Meara; ebs3; Epidermolysis bullosa simplex; k14; Keratin 14; Keratin type I cytoskeletal 14; Keratin14; Koebner; Krt 14; krt14; EBS3; EBS4; NFJ; Keratin, type I cytoskeletal 14; K1C14_HUMAN. Research Area Tumour Cell biology immunology Immunogen Species Mouse Clonality Monoclonal Clone NO. 4A1 React Species Human, Applications IHC-P= IHC-F= ICC= IF= (Paraffin sections need antigen repair)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.Theoretical molecular weight 52kDa Cellular localization cytoplasmic Form Liquid immunogen human CK14 Lsotype IgG1 Purification affinity purified by Protein G Buffer Solution 0.01M PBS(pH7.4) with 1% BSA and 0.1% Proclin300 Storage Store at 2-8 °C for one year. Avoid repeated freeze/thaw cycles. Attention This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. PubMed PubMed Product Detail This gene encodes a member of the keratin family, the most diverse group of intermediate filaments. This gene product, a type I keratin, is usually found as a heterotetramer with two keratin 5 molecules, a type II keratin. Together they form the cytoskeleton of epithelial cells. Mutations in the genes for these keratins are associated with epidermolysis bullosa simplex. At least one pseudogene has been identified at 17p12-p11. [provided by RefSeq].
Function:
The nonhelical tail domain is involved in promoting KRT5-KRT14 filaments to self-organize into large bundles and enhances the mechanical properties involved in resilience of keratin intermediate filaments in vitro.
Subunit:
Heterotetramer of two type I and two type II keratins. keratin-14 associates with keratin-5. Interacts with TRADD and with keratin filaments. Associates with other type I keratins.
Subcellular Location:
Cytoplasm. Nucleus. Note=Expressed in both as a filamentous pattern.
Tissue Specificity:
Detected in the basal layer, lowered within the more apically located layers specifically in the stratum spinosum, stratum granulosum but is not detected in stratum corneum. Strongly expressed in the outer root sheath of anagen follicles but not in the germinative matrix, inner root sheath or hair. Found in keratinocytes surrounding the club hair during telogen.
Post-translational modifications:
A disulfide bond is formed between rather than within filaments and promotes the formation of a keratin filament cage around the nucleus.
DISEASE:
Epidermolysis bullosa simplex, Dowling-Meara type (DM-EBS) [MIM:131760]: A severe form of intraepidermal epidermolysis bullosa characterized by generalized herpetiform blistering, milia formation, dystrophic nails, and mucous membrane involvement. Note=The disease is caused by mutations affecting the gene represented in this entry.
Epidermolysis bullosa simplex, Weber-Cockayne type (WC-EBS) [MIM:131800]: A form of intraepidermal epidermolysis bullosa characterized by blistering limited to palmar and plantar areas of the skin. Note=The disease is caused by mutations affecting the gene represented in this entry. Epidermolysis bullosa simplex, Koebner type (K-EBS) [MIM:131900]: A form of intraepidermal epidermolysis bullosa characterized by generalized skin blistering. The phenotype is not fundamentally distinct from the Dowling-Meara type, although it is less severe. Note=The disease is caused by mutations affecting the gene represented in this entry.
Epidermolysis bullosa simplex, autosomal recessive (AREBS) [MIM:601001]: An intraepidermal epidermolysis bullosa characterized by localized blistering on the dorsal, lateral and plantar surfaces of the feet. Note=The disease is caused by mutations affecting the gene represented in this entry.
Naegeli-Franceschetti-Jadassohn syndrome (NFJS) [MIM:161000]: A rare autosomal dominant form of ectodermal dysplasia. The cardinal features are absence of dermatoglyphics (fingerprints), reticular cutaneous hyperpigmentation (starting at about the age of 2 years without a preceding inflammatory stage), palmoplantar keratoderma, hypohidrosis with diminished sweat gland function and discomfort provoked by heat, nail dystrophy, and tooth enamel defects. Note=The disease is caused by mutations affecting the gene represented in this entry.
Dermatopathia pigmentosa reticularis (DPR) [MIM:125595]: A rare ectodermal dysplasia characterized by lifelong persistent reticulate hyperpigmentation, non-cicatricial alopecia, and nail dystrophy. Variable features include adermatoglyphia, hypohidrosis or hyperhidrosis, and palmoplantar hyperkeratosis. Note=The disease is caused by mutations affecting the gene represented in this entry.
Similarity:
Belongs to the intermediate filament family.
SWISS:
P02533
Gene ID:
3861
Database links:Entrez Gene: 3861 Human
Entrez Gene: 16664 Mouse
Omim: 148066 Human
SwissProt: P02533 Human
SwissProt: Q61781 Mouse
Unigene: 654380 Human
Unigene: 439898 Mouse
Product Picture  Paraformaldehyde-fixed, paraffin embedded (human skin cancer l); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (CK14) Monoclonal Antibody, Unconjugated (BH0073) overnight at 4°C, followed by operating according to SP Kit(Mouse)(sp-0024) instructionsand DAB staining.
Paraformaldehyde-fixed, paraffin embedded (human skin cancer l); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (CK14) Monoclonal Antibody, Unconjugated (BH0073) overnight at 4°C, followed by operating according to SP Kit(Mouse)(sp-0024) instructionsand DAB staining.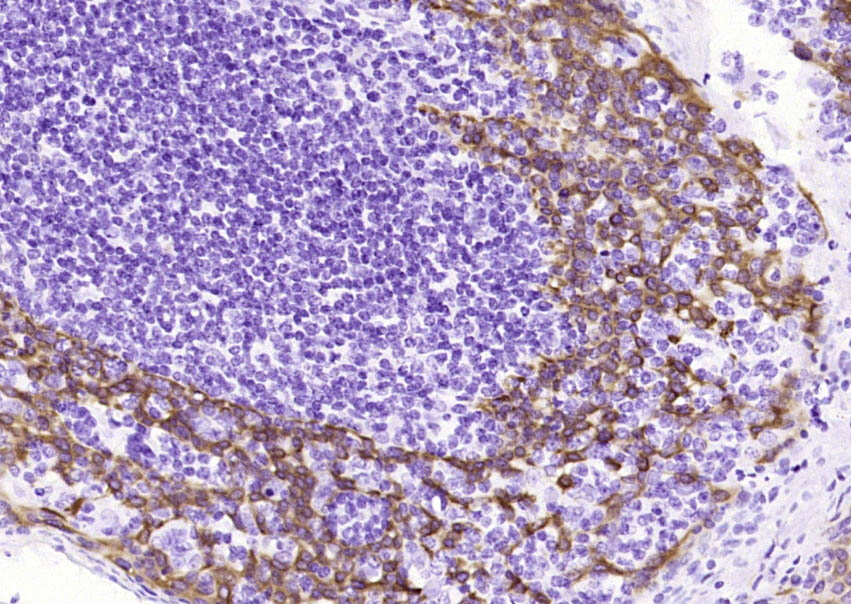 Paraformaldehyde-fixed, paraffin embedded (human tonsil); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (CK14) Monoclonal Antibody, Unconjugated (BH0073) overnight at 4°C, followed by operating according to SP Kit(Mouse)(sp-0024) instructionsand DAB staining.
Paraformaldehyde-fixed, paraffin embedded (human tonsil); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (CK14) Monoclonal Antibody, Unconjugated (BH0073) overnight at 4°C, followed by operating according to SP Kit(Mouse)(sp-0024) instructionsand DAB staining.
Cartpieces
Totalgoods,subtotals:¥Checkout
Partial purchase records(bought amounts latest0)
No one bought this product
User Comment(Total0User Comment Num)
- No comment


 +86 571 56623320
+86 571 56623320




