
Rabbit Anti-beta glucuronidase antibody
asd; Beta G1; Beta glucuronidase; Beta-G1; Beta-glucuronidase; BG; BGLR; BGLR_HUMAN; Glucuronidase beta; Gur; Gus; Gus-r; Gus-s; Gus-t; Gus-u; GUSB; Gut; MPS7; Ac2-223.
View History [Clear]
Details
Product Name beta glucuronidase Chinese Name β葡萄糖醛酸苷酶抗体 Alias asd; Beta G1; Beta glucuronidase; Beta-G1; Beta-glucuronidase; BG; BGLR; BGLR_HUMAN; Glucuronidase beta; Gur; Gus; Gus-r; Gus-s; Gus-t; Gus-u; GUSB; Gut; MPS7; Ac2-223. Research Area Cell biology immunology Signal transduction Immunogen Species Rabbit Clonality Polyclonal React Species Mouse, (predicted: Human, Rat, Chicken, Dog, Pig, Cow, ) Applications WB=1:500-2000 ELISA=1:5000-10000 IHC-P=1:100-500 IHC-F=1:100-500 IF=1:100-500 (Paraffin sections need antigen repair)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.Theoretical molecular weight 69kDa Cellular localization cytoplasmic Form Liquid Concentration 1mg/ml immunogen KLH conjugated synthetic peptide derived from human GUSB/beta glucuronidase: 589-651/651 Lsotype IgG Purification affinity purified by Protein A Buffer Solution 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Storage Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. Attention This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. PubMed PubMed Product Detail Defects in GUSB are the cause of mucopolysaccharidosis type 7 (MPS7) ; also known as Sly syndrome. MPS7 is an autosomal recessive lysosomal storage disease characterized by inability to degrade glucuronic acid-containing glycosaminoglycans. The phenotype is highly variable, ranging from severe lethal hydrops fetalis to mild forms with survival into adulthood. Most patients with the intermediate phenotype show hepatomegaly, skeletal anomalies, coarse facies, and variable degrees of mental impairment. Mucopolysaccharidosis type 7 is associated with non-immune hydrops fetalis, a generalized edema of the fetus with fluid accumulation in the body cavities due to non-immune causes. Non-immune hydrops fetalis is not a diagnosis in itself but a symptom, a feature of many genetic disorders, and the end-stage of a wide variety of disorders.
Function:
Plays an important role in the degradation of dermatan and keratan sulfates.
Subunit:
Homotetramer.
Subcellular Location:
Lysosome.
Post-translational modifications:
N-linked glycosylated with 3 to 4 oligosaccharide chains.
DISEASE:
Defects in GUSB are the cause of mucopolysaccharidosis type 7 (MPS7) [MIM:253220]; also known as Sly syndrome. MPS7 is an autosomal recessive lysosomal storage disease characterized by inability to degrade glucuronic acid-containing glycosaminoglycans. The phenotype is highly variable, ranging from severe lethal hydrops fetalis to mild forms with survival into adulthood. Most patients with the intermediate phenotype show hepatomegaly, skeletal anomalies, coarse facies, and variable degrees of mental impairment.
Note=Mucopolysaccharidosis type 7 is associated with non-immune hydrops fetalis, a generalized edema of the fetus with fluid accumulation in the body cavities due to non-immune causes. Non-immune hydrops fetalis is not a diagnosis in itself but a symptom, a feature of many genetic disorders, and the end-stage of a wide variety of disorders.
Similarity:
Belongs to the glycosyl hydrolase 2 family.
SWISS:
P08236
Gene ID:
2990
Database links:
Entrez Gene: 2990 Human
Omim: 611499 Human
SwissProt: P08236 Human
Unigene: 255230 Human
Product Picture 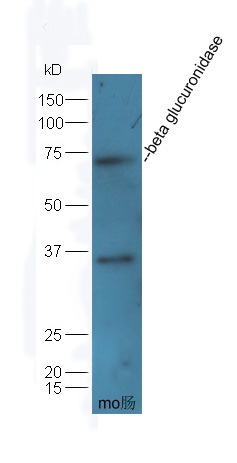 Sample: Intestine (Mouse) Lysate at 40 ug
Sample: Intestine (Mouse) Lysate at 40 ug
Primary: Anti-beta glucuronidase (SL7980R) at 1/300 dilution
Secondary: HRP conjugated Goat-Anti-rabbit IgG (SL0295G-HRP) at 1/5000 dilution
Predicted band size: 69 kD
Observed band size: 69 kD
Cartpieces
Totalgoods,subtotals:¥Checkout
References (0)
No References
Bought notes(bought amounts latest0)
No one bought this product
User Comment(Total0User Comment Num)
- No comment


 +86 571 56623320
+86 571 56623320
 +86 18668110335
+86 18668110335

