
Rabbit Anti-phospho-DAG1 (Tyr892)antibody
DAG1 (phospho-Tyr892); DAG1 (phospho-Y892); p-DAG1(Tyr892); Alpha Dystroglycan (phospho Y892); AGRNR; Alpha-DG; Beta-DG; Beta-dystroglycan; beta Dystroglycan; DAG; Dag1; DAG1_HUMAN; Dystroglycan 1 (dystrophin-associated glycoprotein 1); Dystroglycan; Dyst
View History [Clear]

Details
Product Name phospho-DAG1 (Tyr892) Chinese Name 磷酸化肌萎缩相关蛋白DAG1抗体 Alias DAG1 (phospho-Tyr892); DAG1 (phospho-Y892); p-DAG1(Tyr892); Alpha Dystroglycan (phospho Y892); AGRNR; Alpha-DG; Beta-DG; Beta-dystroglycan; beta Dystroglycan; DAG; Dag1; DAG1_HUMAN; Dystroglycan 1 (dystrophin-associated glycoprotein 1); Dystroglycan; Dystrophin-associated glycoprotein 1; 156DAG; A3a; Dystrophin-associated glycoprotein 1. Product Type Phosphorylated anti Research Area immunology Neurobiology transcriptional regulatory factor Immunogen Species Rabbit Clonality Polyclonal React Species Rat, (predicted: Human, Mouse, Pig, Cow, Horse, Rabbit, Guinea Pig, ) Applications WB=1:500-2000 ELISA=1:5000-10000 IHC-P=1:100-500 IHC-F=1:100-500 IF=1:100-500 (Paraffin sections need antigen repair)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.Theoretical molecular weight 43kDa Cellular localization The nucleus cytoplasmic The cell membrane Extracellular matrix Secretory protein Form Liquid Concentration 1mg/ml immunogen KLH conjugated Synthesised phosphopeptide derived from human DAG1 around the phosphorylation site of Tyr892: PP(p-Y)VP Lsotype IgG Purification affinity purified by Protein A Buffer Solution 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Storage Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. Attention This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. PubMed PubMed Product Detail Dystroglycan is a laminin binding component of the dystrophin-glycoprotein complex which provides a linkage between the subsarcolemmal cytoskeleton and the extracellular matrix. Dystroglycan 1 is a candidate gene for the site of the mutation in autosomal recessive muscular dystrophies. The dramatic reduction of dystroglycan 1 in Duchenne muscular dystrophy leads to a loss of linkage between the sarcolemma and extracellular matrix, rendering muscle fibers more susceptible to necrosis. Dystroglycan also functions as dual receptor for agrin and laminin-2 in the Schwann cell membrane. The muscle and nonmuscle isoforms of dystroglycan differ by carbohydrate moieties but not protein sequence. Alternative splicing results in multiple transcript variants all encoding the same protein.[provided by RefSeq, Apr 2010]
Function:
The dystroglycan complex is involved in a number of processes including laminin and basement membrane assembly, sarcolemmal stability, cell survival, peripheral nerve myelination, nodal structure, cell migration, and epithelial polarization.
Alpha-dystroglycan is an extracellular peripheral glycoprotein that acts as a receptor for both extracellular matrix proteins containing laminin-G domains, and for certain adenoviruses. Receptor for laminin-2 (LAMA2) and agrin in peripheral nerve Schwann cells. Also acts as a receptor for M.leprae in peripheral nerve Schwann cells but only in the presence of the G-domain of LAMA2, and for lymphocytic choriomeningitis virus, Old World Lassa fever virus, and clade C New World arenaviruses.
Beta-dystroglycan is a transmembrane protein that plays important roles in connecting the extracellular matrix to the cytoskeleton. Acts as a cell adhesion receptor in both muscle and non-muscle tissues. Receptor for both DMD and UTRN and, through these interactions, scaffolds axin to the cytoskeleton. Also functions in cell adhesion-mediated signaling and implicated in cell polarity.
Subunit:
Monomer. Heterodimer of alpha- and beta-dystroglycan subunits which are the central components of the dystrophin-glycoprotein complex.
Subcellular Location:
Alpha-dystroglycan: Secreted, extracellular space.Beta-dystroglycan: Cell membrane; Cytoplasm, cytoskeleton. Nucleus, nucleoplasm.
Tissue Specificity:
Expressed in a variety of fetal and adult tissues. In epidermal tissue, located to the basement membrane. Also expressed in keratinocytes and fibroblasts.
Post-translational modifications:
O- and N-glycosylated. Alpha-dystroglycan is heavily O-glycosylated comprising of up to two thirds of its mass and the carbohydrate composition differs depending on tissue type. Mucin-type O-glycosylation is important for ligand binding activity. O-mannosylation of alpha-DAG1 is found in high abundance in both brain and muscle where the most abundant glycan is Sia-alpha-2-3-Gal-beta-1-4-Glc-NAc-beta-1-2-Man. In muscle, glycosylation on Thr-379 by a phosphorylated O-mannosyl glycan with the structure 2-(N-acetylamido)-2-deoxygalactosyl-beta-1,3-2-(N-acetylamido)-2-deoxyglucosyl-beta-1,4-6-phosphomannose is mediated by like-acetylglucosaminyltransferase (LARGE) protein and is required for laminin binding. O-mannosylation is also required for binding lymphocytic choriomeningitis virus, Old World Lassa fever virus, and clade C New World arenaviruses. The O-glycosyl hexose on Thr-367, Thr-369, Thr-372, Thr-381 and Thr-388 is probably mannose. O-glycosylated in the N-terminal region with a core 1 or possibly core 8 glycan. The beta subunit is N-glycosylated. Autolytic cleavage produces the alpha and beta subunits. In cutaneous cells, as well as in certain pathological conditions, shedding of beta-dystroglcan can occur releasing a peptide of about 30 kDa. SRC-mediated phosphorylation of the PPXY motif of the beta subunit recruits SH2 domain-containing proteins, but inhibits binding to WWW domain-containing proteins, DMD and UTRN. This phosphorylation also inhibits nuclear entry.
DISEASE:
Defects in DAG1 are the cause of muscular dystrophy-dystroglycanopathy limb-girdle type C7 (MDDGC7) [MIM:613818]. An autosomal recessive muscular dystrophy showing onset in early childhood, and associated with mental retardation without structural brain anomalies. Note=MDDGC7 is caused by DAG1 mutations that interfere with normal post-translational processing, resulting in defective DAG1 glycosylation and impaired interactions with extracellular-matrix components. Other muscular dystrophy-dystroglycanopathies are caused by defects in enzymes involved in protein O-glycosylation.
Similarity:
Contains 1 peptidase S72 domain.
SWISS:
Q14118
Gene ID:
1605
Database links:Entrez Gene: 1605 Human
Entrez Gene: 13138 Mouse
Omim: 128239 Human
SwissProt: Q14118 Human
SwissProt: Q62165 Mouse
Unigene: 76111 Human
Unigene: 7524 Mouse
Unigene: 36260 Rat
Product Picture 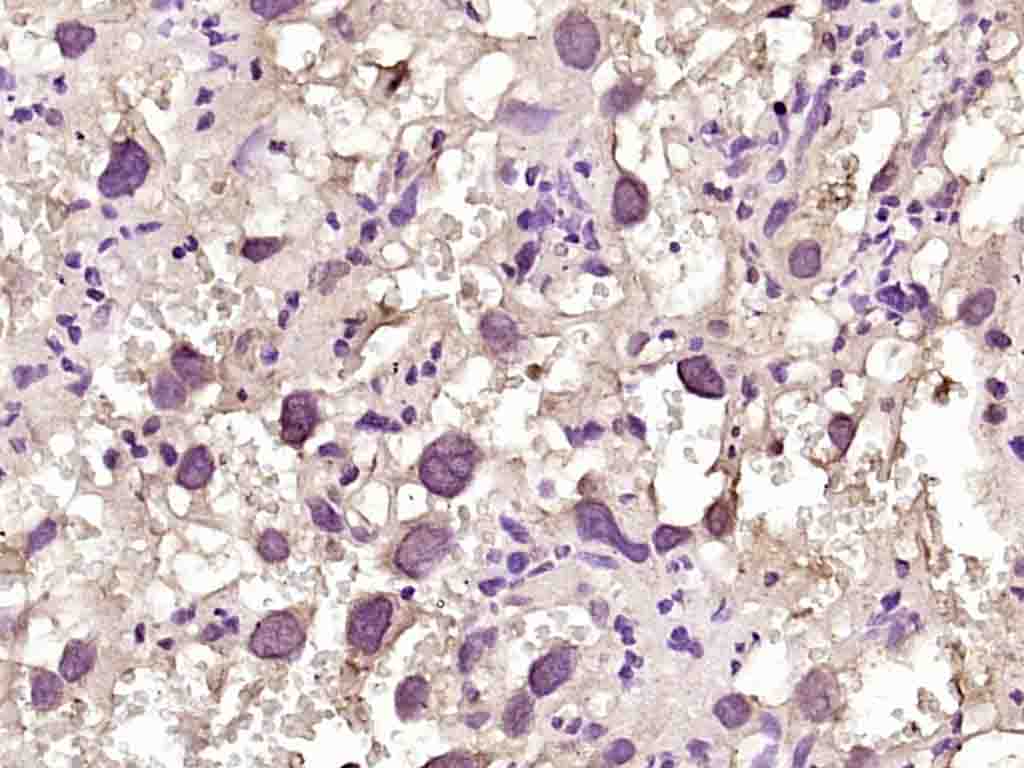 Paraformaldehyde-fixed, paraffin embedded (rat placenta); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (phospho-DAG1 (Tyr892)) Polyclonal Antibody, Unconjugated (SL4075R) at 1:200 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.
Paraformaldehyde-fixed, paraffin embedded (rat placenta); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (phospho-DAG1 (Tyr892)) Polyclonal Antibody, Unconjugated (SL4075R) at 1:200 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.
Cartpieces
Totalgoods,subtotals:¥Checkout
Bought notes(bought amounts latest0)
No one bought this product
User Comment(Total0User Comment Num)
- No comment


 +86 571 56623320
+86 571 56623320
 +86 18668110335
+86 18668110335

