
Rabbit Anti-PSAP antibody
Prosaposin; A1 activator; Cerebroside sulfate activator; Co-beta-glucosidase; Component C; CSAct; Dispersin; GLBA; Glucosylceramidase activator; Proactivator polypeptide; Proactivator polypeptide precursor; Prosaposin (sphingolipid activator protein 1); p
View History [Clear]
Details
Product Name PSAP Chinese Name 鞘脂激活蛋白原抗体 Alias Prosaposin; A1 activator; Cerebroside sulfate activator; Co-beta-glucosidase; Component C; CSAct; Dispersin; GLBA; Glucosylceramidase activator; Proactivator polypeptide; Proactivator polypeptide precursor; Prosaposin (sphingolipid activator protein 1); prosaposin (variant Gaucher disease and variant metachromatic leukodystrophy); Protein A; Protein C; PSAP; SAP-1; SAP-2; SAP_HUMAN; SAP1; Saposin A; Saposin B; Saposin B Val; Saposin C; Saposin D; Saposin-D; Saposins; Sgp1; Sphingolipid activator protein 1; Sphingolipid activator protein 2; Sulfated glycoprotein 1; Sulfatide/GM1 activator. Research Area Tumour immunology Neurobiology Signal transduction Lipoprotein The new supersedes the old Immunogen Species Rabbit Clonality Polyclonal React Species Human, Applications WB=1:500-2000 ELISA=1:5000-10000 IHC-P=1:100-500 IHC-F=1:100-500 IF=1:100-500 (Paraffin sections need antigen repair)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.Theoretical molecular weight 58kDa Cellular localization cytoplasmic Form Liquid Concentration 1mg/ml immunogen KLH conjugated synthetic peptide derived from human Prosaposin: 421-524/524 Lsotype IgG Purification affinity purified by Protein A Buffer Solution 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Storage Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. Attention This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. PubMed PubMed Product Detail This gene encodes a highly conserved glycoprotein which is a precursor for 4 cleavage products: saposins A, B, C, and D. Each domain of the precursor protein is approximately 80 amino acid residues long with nearly identical placement of cysteine residues and glycosylation sites. Saposins A-D localize primarily to the lysosomal compartment where they facilitate the catabolism of glycosphingolipids with short oligosaccharide groups. The precursor protein exists both as a secretory protein and as an integral membrane protein and has neurotrophic activities. Mutations in this gene have been associated with Gaucher disease, Tay-Sachs disease, and metachromatic leukodystrophy. Alternative splicing results in multiple transcript variants encoding different isoforms. [provided by RefSeq, Jul 2008]
Function:
The lysosomal degradation of sphingolipids takes place by the sequential action of specific hydrolases. Some of these enzymes require specific low-molecular mass, non-enzymic proteins: the sphingolipids activator proteins (coproteins).
Saposin-A and saposin-C stimulate the hydrolysis of glucosylceramide by beta-glucosylceramidase (EC 3.2.1.45) and galactosylceramide by beta-galactosylceramidase (EC 3.2.1.46). Saposin-C apparently acts by combining with the enzyme and acidic lipid to form an activated complex, rather than by solubilizing the substrate.
Saposin-B stimulates the hydrolysis of galacto-cerebroside sulfate by arylsulfatase A (EC 3.1.6.8), GM1 gangliosides by beta-galactosidase (EC 3.2.1.23) and globotriaosylceramide by alpha-galactosidase A (EC 3.2.1.22). Saposin-B forms a solubilizing complex with the substrates of the sphingolipid hydrolases.
Saposin-D is a specific sphingomyelin phosphodiesterase activator (EC 3.1.4.12).
Subunit:
Saposin-B is a homodimer.
Subcellular Location:
Lysosome.
Post-translational modifications:
This precursor is proteolytically processed to 4 small peptides, which are similar to each other and are sphingolipid hydrolase activator proteins.
N-linked glycans show a high degree of microheterogeneity.
The one residue extended Saposin-B-Val is only found in 5% of the chains.
DISEASE:
Defects in PSAP are the cause of combined saposin deficiency (CSAPD) [MIM:611721]; also known as prosaposin deficiency. CSAPD is due to absence of all saposins, leading to a fatal storage disorder with hepatosplenomegaly and severe neurological involvement.
Defects in PSAP saposin-B region are the cause of leukodystrophy metachromatic due to saposin-B deficiency (MLD-SAPB) [MIM:249900]. MLD-SAPB is an atypical form of metachromatic leukodystrophy. It is characterized by tissue accumulation of cerebroside-3-sulfate, demyelination, periventricular white matter abnormalities, peripheral neuropathy. Additional neurological features include dysarthria, ataxic gait, psychomotr regression, seizures, cognitive decline and spastic quadriparesis.
Defects in PSAP saposin-C region are the cause of atypical Gaucher disease (AGD) [MIM:610539]. Affected individuals have marked glucosylceramide accumulation in the spleen without having a deficiency of glucosylceramide-beta glucosidase characteristic of classic Gaucher disease, a lysosomal storage disorder.
Defects in PSAP saposin-A region are the cause of atypical Krabbe disease (AKRD) [MIM:611722]. AKRD is a disorder of galactosylceramide metabolism. AKRD features include progressive encephalopathy and abnormal myelination in the cerebral white matter resembling Krabbe disease.
Note=Defects in PSAP saposin-D region are found in a variant of Tay-Sachs disease (GM2-gangliosidosis).
Similarity:
Contains 2 saposin A-type domains.
Contains 4 saposin B-type domains.
SWISS:
P07602
Gene ID:
5660
Database links:Entrez Gene: 5660 Human
Entrez Gene: 19156 Mouse
Omim: 176801 Human
SwissProt: P07602 Human
SwissProt: Q61207 Mouse
Unigene: 523004 Human
Unigene: 277498 Mouse
Unigene: 97173 Rat
Product Picture 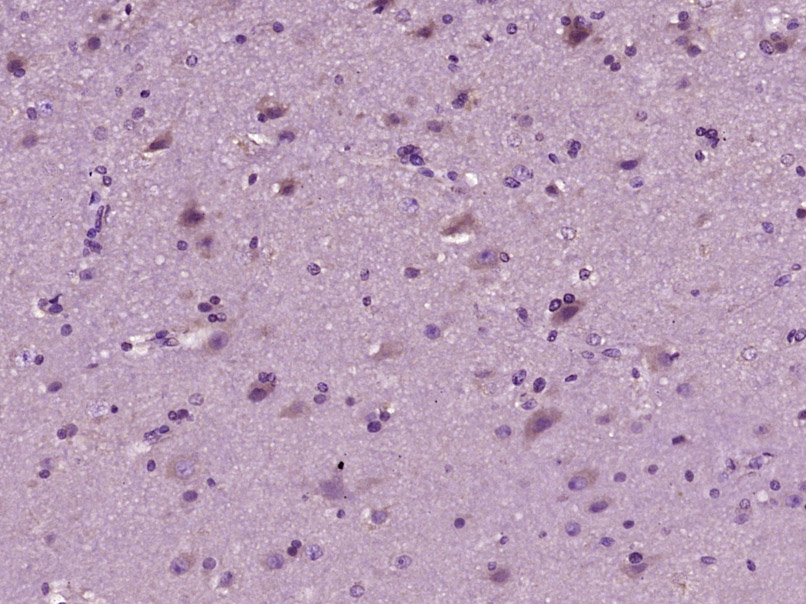 Paraformaldehyde-fixed, paraffin embedded (Human brain glioma); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (PSAP) Polyclonal Antibody, Unconjugated (SL2241R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.
Paraformaldehyde-fixed, paraffin embedded (Human brain glioma); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (PSAP) Polyclonal Antibody, Unconjugated (SL2241R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.
Cartpieces
Totalgoods,subtotals:¥Checkout
References (0)
No References
Bought notes(bought amounts latest0)
No one bought this product
User Comment(Total0User Comment Num)
- No comment


 +86 571 56623320
+86 571 56623320
 +86 18668110335
+86 18668110335

