
Rabbit Anti-TTC21B antibody
ATD4; JBTS11; Nbla10696; NPHP12; Putative protein product of Nbla10696; Tetratricopeptide repeat protein 21B; THM1; TPR repeat protein 21B; TT21B_HUMAN; Ttc21b.
View History [Clear]
Details
Product Name TTC21B Chinese Name 四聚体多肽蛋白21B抗体 Alias ATD4; JBTS11; Nbla10696; NPHP12; Putative protein product of Nbla10696; Tetratricopeptide repeat protein 21B; THM1; TPR repeat protein 21B; TT21B_HUMAN; Ttc21b. Research Area Cell biology Developmental biology Neurobiology Signal transduction Stem cells Immunogen Species Rabbit Clonality Polyclonal React Species Rat, (predicted: Human, Mouse, Dog, Pig, Cow, Horse, Rabbit, Sheep, ) Applications ELISA=1:5000-10000 IHC-P=1:100-500 IHC-F=1:100-500 ICC=1:100-500 IF=1:100-500 (Paraffin sections need antigen repair)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.Theoretical molecular weight 150kDa Cellular localization cytoplasmic Form Liquid Concentration 1mg/ml immunogen KLH conjugated synthetic peptide derived from human TTC21B: 1221-1316/1316 Lsotype IgG Purification affinity purified by Protein A Buffer Solution 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Storage Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. Attention This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. PubMed PubMed Product Detail This gene encodes a member of TTC21 family, containing several tetratricopeptide repeat (TPR) domains. This protein is localized to the cilium axoneme, and may play a role in retrograde intraflagellar transport in cilia. Mutations in this gene are associated with various ciliopathies, nephronophthisis 12, and asphyxiating thoracic dystrophy 4. [provided by RefSeq, Oct 2011]
Function:
May negatively modulate SHH signal transduction and may play a role in retrograde intraflagellar transport in cilia.
Subcellular Location:
Cytoplasm.
DISEASE:
Note=Ciliary dysfunction leads to a broad spectrum of disorders, collectively termed ciliopathies. Overlapping clinical features include retinal degeneration, renal cystic disease, skeletal abnormalities, fibrosis of various organ, and a complex range of anatomical and functional defects of the central and peripheral nervous system. The ciliopathy range of diseases includes Meckel-Gruber syndrome, Bardet-Biedl syndrome, Joubert syndrome, nephronophtisis, Senior-Loken syndrome, and Jeune asphyxiating thoracic dystrophy among others. TTC21B is causally associated with diverse ciliopathies, and also acts as a modifier gene across the ciliopathy spectrum. TTC21B mutations interact in trans with mutations in other ciliopathy-causing genes and contribute to disease manifestation and severity.
Defects in TTC21B are the cause of nephronophthisis type 12 (NPHP12) [MIM:613820]. NPHP12 is an autosomal recessive disorder resulting in end-stage renal disease. It is a progressive tubulo-interstitial kidney disorder histologically characterized by modifications of the tubules with thickening of the basement membrane, interstitial fibrosis and, in the advanced stages, medullary cysts. Some patients manifest extra-renal features including retinal, skeletal and central nervous system defects.
Defects in TTC21B are the cause of asphyxiating thoracic dystrophy type 4 (ATD4) [MIM:613819]. ATD4 is an autosomal recessive chondrodysplasia characterized by a severely constricted thoracic cage, short-limbed short stature, and polydactyly. It often leads to death in infancy because of respiratory insufficiency. Retinal degeneration, cystic renal disease and hepatic disease can be present in affected individuals who survive early childhood.
Defects in TTC21B may be a cause of Bardet-Biedl syndrome (BBS) [MIM:209900]. A syndrome characterized by usually severe pigmentary retinopathy, early-onset obesity, polydactyly, hypogenitalism, renal malformation and mental retardation. Secondary features include diabetes mellitus, hypertension and congenital heart disease. Bardet-Biedl syndrome inheritance is autosomal recessive, but three mutated alleles (two at one locus, and a third at a second locus) may be required for clinical manifestation of some forms of the disease.
Defects in TTC21B may be a cause of Joubert syndrome (JBTS) [MIM:213300]. A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease.
Similarity:
Belongs to the TTC21 family.
Contains 19 TPR repeats.
SWISS:
Q7Z4L5
Gene ID:
79809
Database links:Entrez Gene: 79809 Human
Omim: 612014 Human
SwissProt: Q7Z4L5 Human
Unigene: 310672 Human
Product Picture 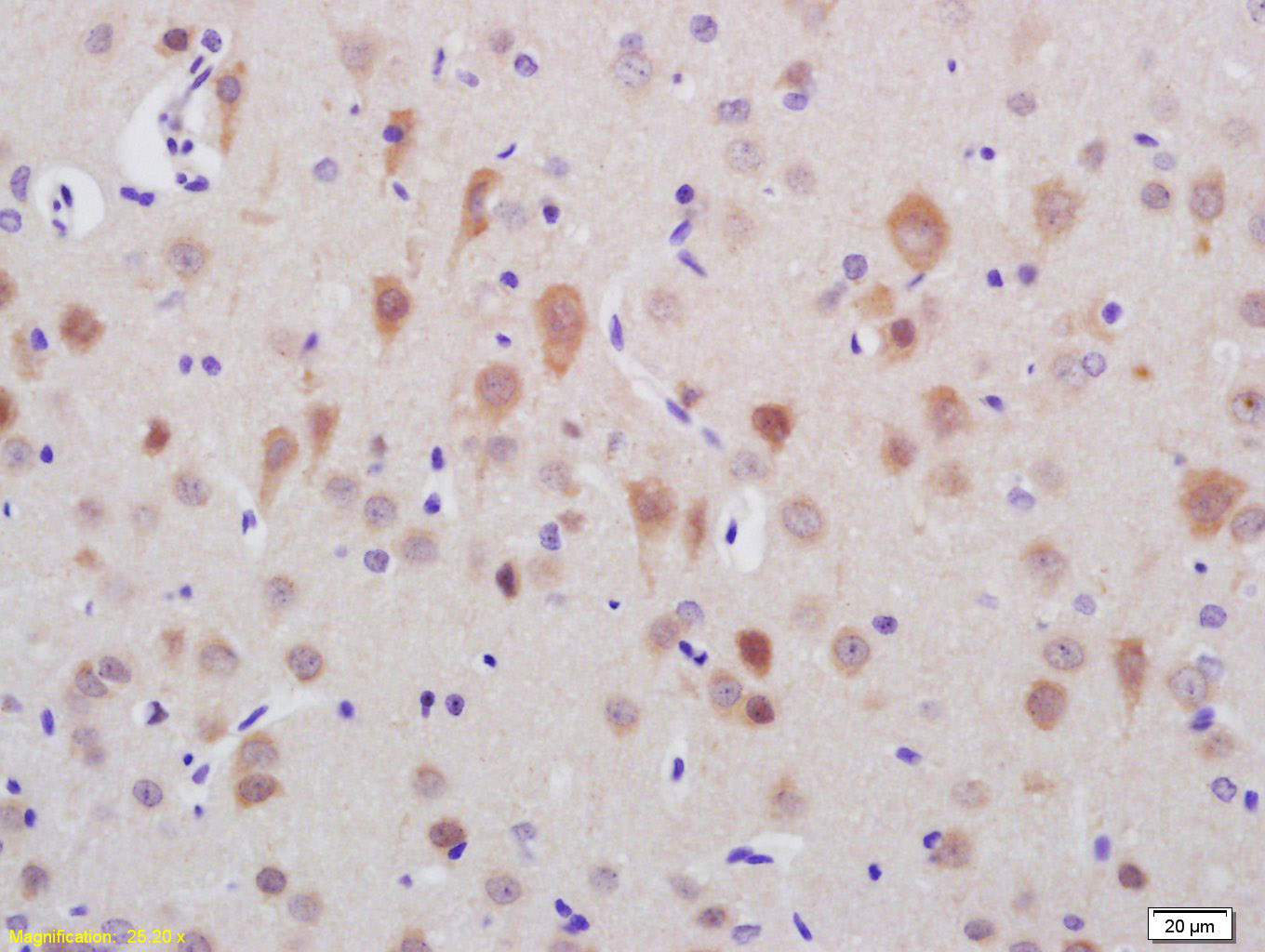 Tissue/cell: rat brain tissue; 4% Paraformaldehyde-fixed and paraffin-embedded;
Tissue/cell: rat brain tissue; 4% Paraformaldehyde-fixed and paraffin-embedded;
Antigen retrieval: citrate buffer ( 0.01M, pH 6.0 ), Boiling bathing for 15min; Block endogenous peroxidase by 3% Hydrogen peroxide for 30min; Blocking buffer (normal goat serum,C-0005) at 37℃ for 20 min;
Incubation: Anti-TTC21B Polyclonal Antibody, Unconjugated(SL12069R) 1:200, overnight at 4°C, followed by conjugation to the secondary antibody(SP-0023) and DAB(C-0010) staining
Cartpieces
Totalgoods,subtotals:¥Checkout
Bought notes(bought amounts latest0)
No one bought this product
User Comment(Total0User Comment Num)
- No comment


 +86 571 56623320
+86 571 56623320
 +86 18668110335
+86 18668110335

