
Rabbit Anti-GBA antibody
Glucosidase beta; Acid beta glucosidase; Acid beta-glucosidase; Alglucerase; Beta glucocerebrosidase; BETA GLUCOSIDASE, ACID; Beta-glucocerebrosidase; betaGC; D glucosyl N acylsphingosine glucohydrolase; D-glucosyl-N-acylsphingosine glucohydrolase; EC 3.2
View History [Clear]
Details
Product Name GBA Chinese Name β-葡萄糖脑苷脂酶抗体 Alias Glucosidase beta; Acid beta glucosidase; Acid beta-glucosidase; Alglucerase; Beta glucocerebrosidase; BETA GLUCOSIDASE, ACID; Beta-glucocerebrosidase; betaGC; D glucosyl N acylsphingosine glucohydrolase; D-glucosyl-N-acylsphingosine glucohydrolase; EC 3.2.1.45 ; Gba protein; GBA1; GC antibody GCase; GCB; GLCM_HUMAN; GLUC; Glucocerebrosidase (alt.); Glucocerebrosidase; GLUCOCEREBROSIDASE PSEUDOGENE; Glucosidase beta; Glucosidase, beta, acid; Glucosidase, beta; acid (includes glucosylceramidase); Glucosylceramidase; Imiglucerase; Lysosomal glucocerebrosidase. Research Area Tumour Cell biology Neurobiology Signal transduction The new supersedes the old Immunogen Species Rabbit Clonality Polyclonal React Species Human, (predicted: Mouse, Rat, Rabbit, ) Applications WB=1:500-2000 ELISA=1:5000-10000 IHC-P=1:100-500 IHC-F=1:100-500 ICC=1:100-500 IF=1:100-500 (Paraffin sections need antigen repair)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.Theoretical molecular weight 56kDa Cellular localization cytoplasmic The cell membrane Form Liquid Concentration 1mg/ml immunogen KLH conjugated synthetic peptide derived from human GBA: 141-240/536 Lsotype IgG Purification affinity purified by Protein A Buffer Solution 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Storage Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. Attention This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. PubMed PubMed Product Detail This gene encodes a lysosomal membrane protein that cleaves the beta-glucosidic linkage of glycosylceramide, an intermediate in glycolipid metabolism. Mutations in this gene cause Gaucher disease, a lysosomal storage disease characterized by an accumulation of glucocerebrosides. A related pseudogene is approximately 12 kb downstream of this gene on chromosome 1. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jan 2010]
Subunit:
Interacts with saposin-C. Interacts with SCARB2.
Subcellular Location:
Lysosome membrane. Interaction with saposin-C promotes membrane association.
DISEASE:
Defects in GBA are the cause of Gaucher disease (GD) [MIM:230800]; also known as glucocerebrosidase deficiency. GD is the most prevalent lysosomal storage disease, characterized by accumulation of glucosylceramide in the reticulo-endothelial system. Different clinical forms are recognized depending on the presence (neuronopathic forms) or absence of central nervous system involvement, severity and age of onset.
Defects in GBA are the cause of Gaucher disease type 1 (GD1) [MIM:230800]; also known as adult non-neuronopathic Gaucher disease. GD1 is characterized by hepatosplenomegaly with consequent anemia and thrombopenia, and bone involvement. The central nervous system is not involved.
Defects in GBA are the cause of Gaucher disease type 2 (GD2) [MIM:230900]; also known as acute neuronopathic Gaucher disease. GD2 is the most severe form and is universally progressive and fatal. It manifests soon after birth, with death generally occurring before patients reach two years of age.
Defects in GBA are the cause of Gaucher disease type 3 (GD3) [MIM:231000]; also known as subacute neuronopathic Gaucher disease. GD3 has central nervous manifestations. Defects in GBA are the cause of Gaucher disease type 3C (GD3C) [MIM:231005]; also known as pseudo-Gaucher disease or Gaucher-like disease.
Defects in GBA are the cause of Gaucher disease perinatal lethal (GDPL) [MIM:608013]. It is a distinct form of Gaucher disease type 2, characterized by fetal onset. Hydrops fetalis, in utero fetal death and neonatal distress are prominent features. When hydrops is absent, neurologic involvement begins in the first week and leads to death within 3 months.
Hepatosplenomegaly is a major sign, and is associated with ichthyosis, arthrogryposis, and facial dysmorphism.
Note=Perinatal lethal Gaucher disease is associated with non-immune hydrops fetalis, a generalized edema of the fetus with fluid accumulation in the body cavities due to non-immune causes. Non-immune hydrops fetalis is not a diagnosis in itself but a symptom, a feature of many genetic disorders, and the end-stage of a wide variety of disorders.
Defects in GBA contribute to susceptibility to Parkinson disease (PARK) [MIM:168600]. A complex neurodegenerative disorder characterized by bradykinesia, resting tremor, muscular rigidity and postural instability. Additional features are characteristic postural abnormalities, dysautonomia, dystonic cramps, and dementia. The pathology of Parkinson disease involves the loss of dopaminergic neurons in the substantia nigra and the presence of Lewy bodies (intraneuronal accumulations of aggregated proteins), in surviving neurons in various areas of the brain. The disease is progressive and usually manifests after the age of 50 years, although early-onset cases (before 50 years) are known. The majority of the cases are sporadic suggesting a multifactorial etiology based on environmental and genetic factors. However, some patients present with a positive family history for the disease. Familial forms of the disease usually begin at earlier ages and are associated with atypical clinical features.
Similarity:
Belongs to the glycosyl hydrolase 30 family.
SWISS:
P04062
Gene ID:
2629
Database links:Entrez Gene: 2629 Human
Entrez Gene: 14466 Mouse
Omim: 606463 Human
SwissProt: P04062 Human
SwissProt: P17439 Mouse
Unigene: 282997 Human
Unigene: 719930 Human
Unigene: 5031 Mouse
Unigene: 162606 Rat
Product Picture 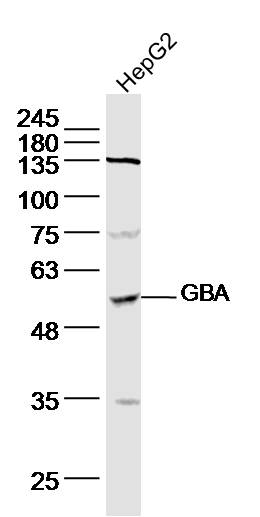 Sample: HepG2(Human) Cell Lysate at 40 ug
Sample: HepG2(Human) Cell Lysate at 40 ug
Primary: Anti-GBA (SL11779R) at 1/300 dilution
Secondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilution
Predicted band size: 56 kD
Observed band size: 56 kD
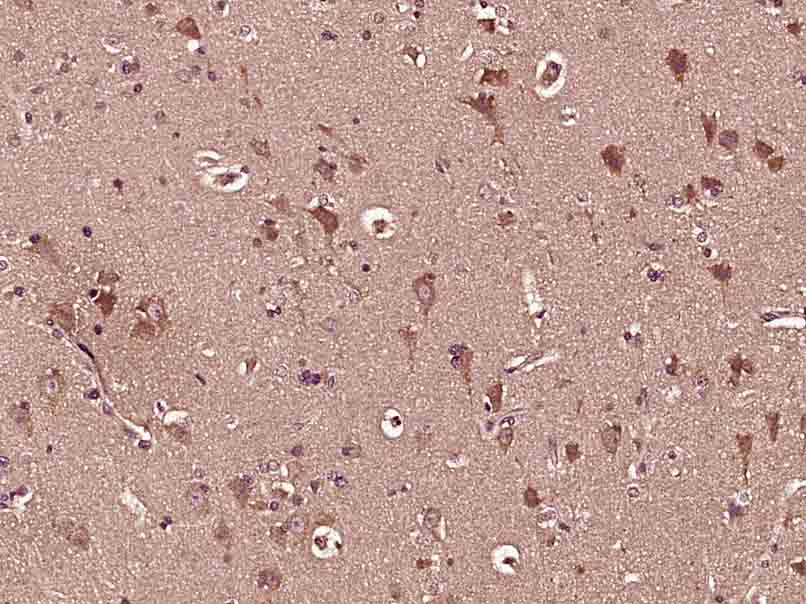 Paraformaldehyde-fixed, paraffin embedded (Human brain glioma); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (GBA) Polyclonal Antibody, Unconjugated (SL11779R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.
Paraformaldehyde-fixed, paraffin embedded (Human brain glioma); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (GBA) Polyclonal Antibody, Unconjugated (SL11779R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.
Cartpieces
Totalgoods,subtotals:¥Checkout
Bought notes(bought amounts latest0)
No one bought this product
User Comment(Total0User Comment Num)
- No comment


 +86 571 56623320
+86 571 56623320
 +86 18668110335
+86 18668110335

