
Rabbit Anti-KCTD7 antibody
BTB/POZ domain containing protein KCTD7; EPM3; FLJ32069; Potassium channel tetramerisation domain containing 7; KCTD7_HUMAN.
View History [Clear]
Details
Product Name KCTD7 Chinese Name 钾离子通道多聚体结构域蛋白7抗体 Alias BTB/POZ domain containing protein KCTD7; EPM3; FLJ32069; Potassium channel tetramerisation domain containing 7; KCTD7_HUMAN. Research Area Cell biology Neurobiology Channel protein The cell membrane受体 Immunogen Species Rabbit Clonality Polyclonal React Species Human, (predicted: Mouse, Rat, Chicken, Dog, Pig, Cow, Horse, Rabbit, Sheep, ) Applications WB=1:500-2000 ELISA=1:5000-10000 IHC-P=1:100-500 IHC-F=1:100-500 Flow-Cyt=1ug/test ICC=1:100-500 IF=1:100-500 (Paraffin sections need antigen repair)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.Theoretical molecular weight 33kDa Cellular localization cytoplasmic The cell membrane Form Liquid Concentration 1mg/ml immunogen KLH conjugated synthetic peptide derived from human KCTD7: 112-180/289 Lsotype IgG Purification affinity purified by Protein A Buffer Solution 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Storage Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. Attention This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. PubMed PubMed Product Detail Epilepsy affects about 0.5% of the world’s population and has a large genetic component. Epilepsy results from an electrical hyperexcitability in the central nervous system. Potassium channels are important regulators of electrical signaling, determining the firing properties and responsiveness of a variety of neurons. Benign familial neonatal convulsions (BFNC), an autosomal dominant epilepsy of infancy, has been shown to be caused by mutations in the KCNQ2 or the KCNQ3 potassium channel genes. KCNQ2 and KCNQ3 are voltage-gated potassium channel proteins with six putative transmembrane domains. Both proteins display a broad distribution within the brain, with expression patterns that largely overlap.
Function:
The KCTD gene family, including KCTD7, encode predicted proteins that contain N terminal domain that is homologous to the T1 domain in voltage gated potassium channels. KCTD7 displays a primary sequence and hydropathy profile indicating intracytoplasmic localization. There are two named isoforms.
Subunit:
May be involved in the control of excitability of cortical neurons
Subcellular Location:
Cell membrane. Cytoplasm, cytosol.
DISEASE:
efects in KCTD7 are the cause of epilepsy, progressive myoclonic 3, with or without intracellular inclusions (EPM3) [MIM:611726]. EPM3 is an autosomal recessive, severe, progressive myoclonic epilepsy with early-onset. Multifocal myoclonic seizures begin between 16 and 24 months of age after normal initial development. Neurodegeneration and regression occur with seizure onset. Other features include mental retardation, dysarthria, truncal ataxia, and loss of fine finger movements. EEG shows slow dysrhythmia, multifocal and occasionally generalized epileptiform discharges. In some patients, ultrastructural findings on skin biopsies identify intracellular accumulation of autofluorescent lipopigment storage material, consistent with neuronal ceroid lipofuscinosis.
Note=Defects in KCTD7 are a cause of opsoclonus-myoclonus ataxia-like syndrome. Opsoclonus myoclonus ataxia syndrome (OMS) is a rare pervasive and frequently permanent disorder that usually develops in previously healthy children with normal premorbid psychomotor development and characterized by association of abnormal eye movements (opsoclonus), severe dyskinesia (myoclonus), cerebellar ataxia, functional regression, and behavioral problems. The syndrome is considered to be an immune-mediated disorder and may be tumor-associated or idiopathic. OMS is one of a few steroid responsive disorders of childhood. KCTD7 mutations have been found in a patient with an atypical clinical presentation characterized by non-epileptic myoclonus and ataxia commencing in early infancy, abnormal opsoclonus-like eye movements, improvement of clinical symptoms under steroid treatment, and subsequent development of generalized epilepsy (PubMed:22638565).
Similarity:
Contains 1 BTB (POZ) domain.
SWISS:
Q96MP8
Gene ID:
154881
Database links:Entrez Gene: 417547 Chicken
Entrez Gene: 154881 Human
Entrez Gene: 212919 Mouse
Omim: 611725 Human
SwissProt: Q5ZJP7 Chicken
SwissProt: Q96MP8 Human
SwissProt: Q8BJK1 Mouse
Unigene: 546627 Human
Unigene: 55812 Mouse
Unigene: 103510 Rat
Product Picture 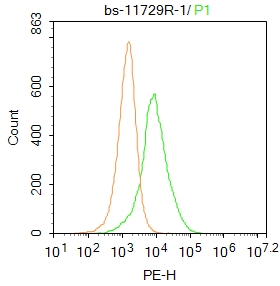 Blank control: Hela.
Blank control: Hela.
Primary Antibody (green line): Rabbit Anti-KCTD7 antibody (SL11729R)
Dilution: 1μg /10^6 cells;
Isotype Control Antibody (orange line): Rabbit IgG .
Secondary Antibody : Goat anti-rabbit IgG-PE
Dilution: 1μg /test.
Protocol
The cells were fixed with 4% PFA (10min at room temperature)and then permeabilized with 20% PBST for 20 min atroom temperature. The cells were then incubated in 5%BSA to block non-specific protein-protein interactions for 30 min at at room temperature .Cells stained with Primary Antibody for 30 min at room temperature. The secondary antibody used for 40 min at room temperature. Acquisition of 20,000 events was performed.
Cartpieces
Totalgoods,subtotals:¥Checkout
Bought notes(bought amounts latest0)
No one bought this product
User Comment(Total0User Comment Num)
- No comment


 +86 571 56623320
+86 571 56623320
 +86 18668110335
+86 18668110335

